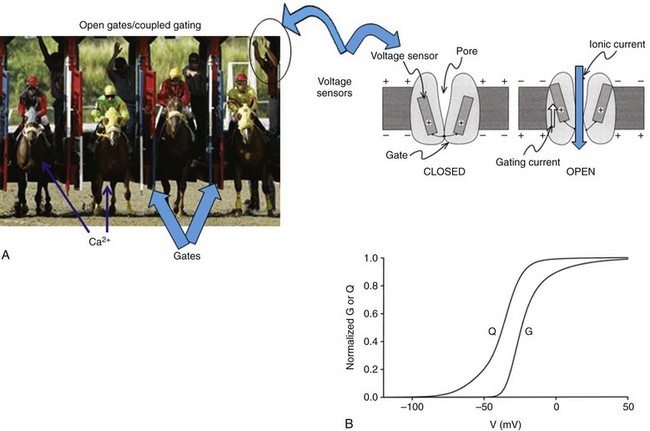
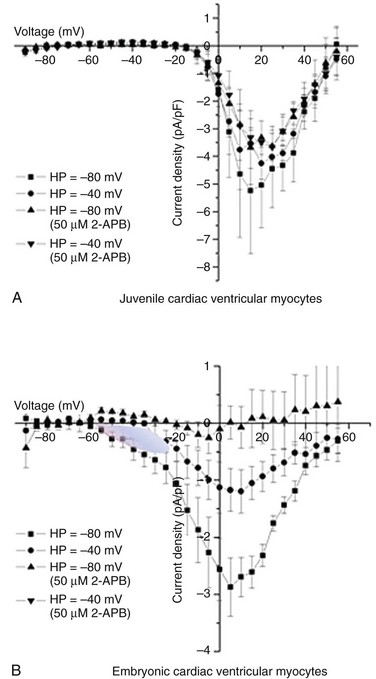
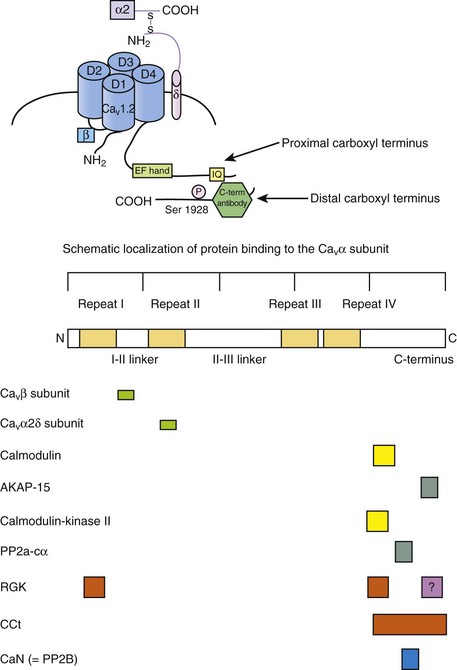
10 The super-family of voltage-gated ion channels is defined by the common ability of the protein to sense transmembrane potential. Voltage-gated ion channels, specifically voltage-gated cation channels, share the general structural plan consisting of six α-helical transmembrane segments and a region of amino acids between transmembrane segments 5 and 6 (S5 and S6) that fold in from the extracellular space toward the cytosol to form the outer permeation pathway. The fundamental common property of voltage sensing is conferred by a transmembrane α-helical stretch of amino acids in the fourth transmembrane segment (S4). The purpose of this chapter is to describe the regulation of cardiac calcium–channel gating. Gating is not simply movement of S4 segments. Additional detail on calcium channels and cardiac myocyte physiology can be gleaned from several excellent reviews,1–3 as well as two outstanding books on ion channel biophysics.4,5 Voltage-gated Ca2+ channels tend to open in response to depolarization and tend to close after repolarization. Channel transiting from an open to a closed state is termed activation gating. Channel gating can be envisioned as a mechanical contraption that allows ionic flow across a barrier (Figure 10-1). It requires some imagination to picture a complex molecule undergoing gating. Consider one of the starting gate slots at the Kentucky Derby. In this analogy, the stationary horse represents the potential energy of a calcium ion affected by its electrochemical gradient. Once the gate opens, the calcium ion flows down its electrochemical gradient. Although the ion may interact with the gate, and influence the gate, the gate is operated independently. Moreover, there are a series of sequential complex steps between the starter pushing the button for the gates to open and the actual gating event. Similarly, initial movement of the S4 in response to voltage is transmitted in a complex, incompletely understood fashion to other domains of the channel that alter the permeation pathway, allowing ionic flux. Figure 10-1 The relationship among voltage sensing, ion channel gating, and conductance. A, Schematic shows the principal features of a generalized voltage-gated ion channel and the metaphorical electric-gated Kentucky Derby starting gate. The α-helical transmembrane segments (S4) sense the electrical gradient. Motion of the S4 segments is transmitted allosterically through the channel protein to open the gate. Ions flux based on their electrochemical gradient. B, Conductance (G) and gating charge movement (Q) depend on voltage for a voltage-dependent ion channel. (B, Adapted from Bezanilla F: The voltage sensor in voltage-dependent ion channels. Physiol Rev 80:555–592, 2000.) Cardiac L-type Ca2+-channel gating is influenced by voltage, Ca2+-ion posttranslational modifications and protein-protein interactions. Voltage is a major determinant of gating, and superimposed effects of permeating cations, as well as modulation of gating primarily by channel phosphorylation status, must be considered. Finally, the cardiac-calcium channel is a heteromultimeric protein complex. Accessory proteins and interacting proteins either directly or via scaffolding proteins modify cardiac calcium–channel gating. Additional details on L-type Ca2+ channel in the heart can be found in Chapter 2, focus on excitation-contraction coupling is covered in Chapter 16, β-adrenergic regulation of cardiac function is reviewed in Chapter 19, and a more in-depth focus on Timothy syndrome is presented in Chapter 94. The myocardium expresses L-type and T-type voltage-gated calcium channels. Myocardial L-type calcium channels include CaV1.2 and CaV1.3. Mature ventricular myocardium almost exclusively expresses the CaV1.2 channel. CaV1.2 and CaV1.3 are expressed in atrial cardiomyocytes, and CaV1.3 is also expressed by sinoatrial6 and atrioventricular nodal cells.7 Commensurate with these tissue localizations, CaV1.2 is critically important for providing trigger calcium for excitation-contraction coupling, and CaV1.3 contributes to heart rate and cardiac conduction.8,9 In the ventricular myocardium, there is an age- and gender-dependent gradient of ICa,L. Prepubertal boys have elevated ICa,L at the base, but developing girls do not. In adult rabbits, the gradients are somewhat reversed, with females displaying higher ICa,L at baseline compared with males,10 and this may be related to estrogen regulation of L-type Ca2+ gene transcription.11 T-type Ca2+ channels in the heart are encoded by CaV3.1 and CaV3.2 pore-forming subunits.12,13 T-type Ca2+ current, ICa,T, is not normally observed in the mature mammalian ventricular myocardium. ICa,T is present in pacemaker cells,14 atrial cells, and Purkinje fibers.15 ICa,T is also expressed in developing cardiomyocytes.13,16,17 Consistent with the adage that pathologic cardiac hypertrophy is accompanied by reexpression of the fetal gene program, ICa,T is reexpressed in ventricular hypertrophy in the cat18 and rat.19 The gating properties of ICa,T versus ICa,L are fundamentally distinct. T-type versus L-type channels are also classified as low voltage–activated versus high voltage–activated, respectively. The activation range of ICa,T is positive to approximately −60 mV, whereas ICa,L activates positive to approximately −20 mV. Closed-state inactivation also follows a similar general pattern. T-type Ca2+ channel steady-state availability is maximal for voltages less than −90 mV, whereas substantial closed-state L-type channel inactivation is not observed for potentials as positive as approximately −40 mV. The positive shift of ICa,L availability (also known as steady-state inactivation) has a practical benefit for measuring ionic current in ventricular cardiomyocytes as well. Voltage-gated Na+ channels are largely inactivated at −40 mV, thus a prepulse to −40 mV is frequently used to isolate ICa,L from prominent overlapping INa. An alternative method used to measure ICa,L in ventricular cardiomyocytes is to replace external Na+ with an impermeant cation. ICa,L can then be measured using more negative holding potentials. In juvenile ventricular cardiomyocytes, hyperpolarizing the holding potential from −40 to −80 mV has only minor effects on peak ICa,L (Figure 10-2, A). By contrast, embryonic ventricular cardiomyocytes exhibit ICa,T and ICa,L. ICa,T manifests itself as a current activating positive to −60 mV when a −80 mV holding potential is used, and this low voltage–activated Ca2+ current (ICa,T) is closed state–inactivated by holding the potential at −40 mV (see Figure 10-2, B). Heterologous expression systems transfected with pore-forming CaV3.x subunits alone reconstitute most of the native ICa,T properties.13 In sharp contrast, native ICa,L gating requires auxiliary proteins and in some cases is rather difficult to completely recreate in heterologous expression systems. The remainder of this chapter will focus on CaV1.2, the predominant L-type Ca2+ channel. Figure 10-2 L-type current (ICa,L) is the only discernible ICa in mature cardiac myocytes. ICa,L and T-type current are simultaneously functional in developing ventricular myocardium. Current-voltage curve for juvenile (1 to 2-month-old mice) (A) and embryonic (B) ventricular myocytes. A, ICa,L without detectable ICa,T manifested as equivalent peak current for holding potentials (Vhold) of −80 and −40 mV. B, ICa,T is elicited from Vhold −80 mV and is steady-state inactivated at Vhold −40, resulting in no low-voltage–activated current detected upon depolarization. The shaded area indicates the ICa,T component; ICa,T also known as low-voltage-activated Ca2+ current is observed between −60 and −20 mV (blue shaded region in panel B). 2-APB does not block CaV1.2 but does inhibit non–CaV1.2-Ca2+ current. The 2-APB sensitivity thus illustrates the complex mixture of T- and multiple L-type currents in the developing myocardium, in contrast to ICa,L dominated by CaV1.2 in mature ventricular cardiomyocytes. (Adapted from Schroder E, Wei Y, Satin J: The developing cardiac myocyte: maturation of excitability and excitation-contraction coupling. Ann N Y Acad Sci 1080:63–75, 2006.) The pore-forming CaV1.2 Ca2+ channel does not gate in isolation in cardiac myocytes, and studies of ionic current in heterologous expression systems show that a functional ion channel complex requires auxiliary proteins. Although the total number of CaV1.2-interacting proteins is unknown, several important interacting partners have been identified and studied (Figure 10-3). Early electrophysiological studies in nonexcitable cells heterologously expressing CaV1.2 alone showed little, if any, discernible ionic current. Coexpression of CaVβ subunits is a requirement to study L-type Ca current from channels generated by plasmids introduced into nonexcitable cells.20 CaVβ–CaV1.2 interactions occur via a deep hydrophobic pocket of CaVβ complexed with the LI-II domain of CaV1.2.21 CaVβ–CaV1.2 interactions have gating and nongating consequences. Early studies showed that CaVβ masks an endoplasmic reticulum retention signal on CaV1.2, thus allowing CaV1.2 to traffic to the surface membrane.22 More recent studies show that CaVβ–CaV1.2 binding influences a CaV1.2 carboxyl-terminal rearrangement to promote surface expression.23 Despite the relatively high-affinity CaV1.2–CaVβ interaction (2-54 nM), dynamic CaVβ–CaV1.2 interaction may also occur with respect to L-type Ca2+-channel gating,24 consistent with the idea that CaVβ modifies CaV1.2 gating in a regulated fashion. Figure 10-3 Subunit structure of L-type Ca2+–channel complex. The upper panel depicts four homologous repeats of pore-forming CaV1.2 subunit, CaVβ bound to LI-II (linker joining repeats I and II), and the proximal carboxyl-terminus. The IQ motif is required for CaM binding. The distal carboxyl-terminus is shown as a proteolytically cleaved protein. Antibodies raised against this segment show it migrating as an independent protein from the pore-forming region. The lower panel summarizes critical interacting proteins along with approximate CaV1.2 interaction domains. Green boxes indicate regulators that tend to increase ICa,L, yellow boxes denote more complex effects, and red boxes inhibit ICa,L. Calcineurin (CaN) is denoted with a blue box because of the controversial effects of CaN on ICa,L. The CaV1.2 carboxyl-terminus spans approximately 300 amino acids (size differing among splice variants), from the cytosolic border of homologous repeat intravenous transmembrane S6 until the termination of the protein. The distal carboxyl-terminus is proteolytically cleaved, yielding an approximate 37 kDa protein that covalently reassociates with the proximal carboxyl-terminus to regulate function25 (also discussed later). The distal carboxyl-terminus also can localize to the nucleus,26 where it regulates gene transcription, including that for CaV1.2.27 The proximal carboxyl-terminus remains contiguous with the pore-forming CaV1.2. Calmodulin (CaM) is prebound to CaV1.2 on the proximal carboxyl-terminus in the IQ motif28,29 and is a critical determinant of Ca2+-dependent inactivation gating (discussed later). IQ is the single letter abbreviation for isoleucine and glutamine. The “I” of the IQ motif is essential for CaM binding,30,31 and mutation of isoleucine of the IQ motif induces dilated cardiomyopathy and premature death.32 Two CaM molecules interact with the proximal carboxyl-terminus of CaV1.2, and the CaM molecules on CaV1.2 are arranged in an antiparallel fashion.31,33 The proximal carboxyl-terminus of CaV1.2 also interacts with several other proteins that modify channel gating. The monomeric G-protein Rem functionally competes with CaM for channel regulation at this domain as well.34 CaMKII tethers to the proximal carboxyl-terminus and is an important modulator of CaV1.2 activity.35 Currently, it is unclear how many proteins combine to form the native L-type calcium channel complex. An unbiased proteomics screen of the closely related N-type calcium channel, CaV2.2, revealed channel interactions with 207 proteins.36 This suggests that multiple protein-protein interactions sum to yield native ICa,L properties. In fact, the number of interacting proteins for the CaV1.2 carboxyl-terminal domain exceeds the restricted space, suggesting that weak protein-protein interactions among multiple proteins create the possibility of diverse mixtures of proteins for any given L-type Ca2+-channel complex. Some of the known CaV1.2 interacting proteins are summarized in Table 10-1. Table 10-1 Inactivation gating is not simply the reverse of activation gating. There is no complete molecular structure data available for voltage-gated Ca channels; however, voltage-gated Na channels have recently been crystallized in two potentially inactivated states.49 These crystal structure studies support biophysical studies that suggest that inactivation gating consists of a series of complex molecular motions, whereby the voltage sensing domains (S4) shift around the pore, two of the S6 segments are transposed extracellularly, and the other two S6 segments collapse on the pore. Consequentially, the permeation pathway is reshaped. Drawing on the conservation of such broad structure-function models, it can be inferred that similarly complex motions impart voltage-dependent inactivation to mammalian voltage-gated calcium channels. The generalized structure of L-type calcium channels (CaV1.x) shares the voltage-gated ion channel super-family plan of six α-helical transmembrane domains, arranged as four contiguous homologous repeats. There are obvious critically important distinctions in structure-function detail between CaV1.x and other voltage-gated ion channels. The CaV1.x processed transcript encodes on the order of more than 2100 amino acids; the precise number depends on splice variant expression. By contrast to T-type Ca2+ channels, and closely related NaV channels, CaV channels require various subunits to generate basal function. Perhaps the single most critical class of subunits are the CaVβ—mainly CaVβ2 in the myocardium.50 CaV1.x and CaVβ form tight interactions. Cytosolic CaVβ increases cell surface expression and increases channel gating.51 Early crystallographic studies identified a hydrophobic groove on CaVβ that confers CaV1 interactions.21,33,52 More recent work sheds light on α–β interactions with respect to CaV1 structures and, in doing so, yields insight into L-type Ca2+-channel gating. More than a decade ago, the cytoplasmic I-II linker (LI-II) of CaV1.2 was identified as essential for CaVβ interaction.53 Subdomains of the cytoplasmic LI-II are highly conserved among CaV1.x channels and across species consistent with conservation of function. CaVβ–LI-II interaction may also contribute to gating. The S6 segment of CaV1.2 is thought to form the inner permeation pathway and lies adjacent to the proximal LI-II of CaV1.2. Thus, crystallography data support the revised model that CaVβ binding transmits changes to inactivation gating of CaV1.2 via a partial α-helical proximal LI-II segment.54
Regulation of Cardiac Calcium Channels
Overview
Calcium Channel Expression in the Myocardium
L-type versus T-type Channel Expression and Gating
The Cardiac L-type Calcium Channel Is a Multiprotein Complex
Interacting Protein
Interaction site on CaV1.2
References
CaV1.2
DCT, N-terminus
37-39
CaVβ2
LI-II, PCT
40
CaV1.2–DCT
PCT
25
α2δ
Uncertain
Calmodulin (CaM)
IQ motif of PCT
See text below
Calmodulin kinase II (CaMKII)
PCT
35
Rem
Via CaVβ2, DCT, N-terminus
34; 41
Rad
Via CaVβ2, N-terminus
41
Calcineurin (CaN = PP2B)
DCT
42; 43
PP2A
DCT
43
PDE4B
CaV1.2 by ip, no subdomain determined
44
Akap150/79
PCT–DCT scaffolds with other proteins
45
PKA
PCT–DCT via akap
46
α-Actinin
PCT/DCT
47
Sorcin
PCT
48
CaV1.2 Structure and Gating
Regulation of Cardiac Calcium Channels



