Fig. 36.1
Hermansky-Pudlak syndrome. CT images through upper and lower lung zones showing extensive subpleural honeycombing bilaterally, more prominent in the lower lung zones where ground glass opacity is also present
Unlike cystic fibrosis, which is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, the genetic of diffuse parenchymal lung disease is complex. For example, idiopathic interstitial pneumonia display considerable genetic heterogeneity and both rare mutations (within surfactant protein C, surfactant protein A2, telomerase reverse transcriptase and telomerase RNA component genes [1–7]) and common variants (MUC5B gene [8]) have been associated with the risk of developing the disease (Fig. 36.2). In familial forms of pulmonary fibrosis many of the pedigrees show vertical transmission consistent with a genetic pattern of autosomal dominant inheritance with reduced penetrance. Interestingly, 45 % of the pedigrees display phenotypic heterogeneity, suggesting that the underlying genetic factors cause an increased generic predisposition for pulmonary fibrosis, and not a predisposition for a specific disease subtype [9].
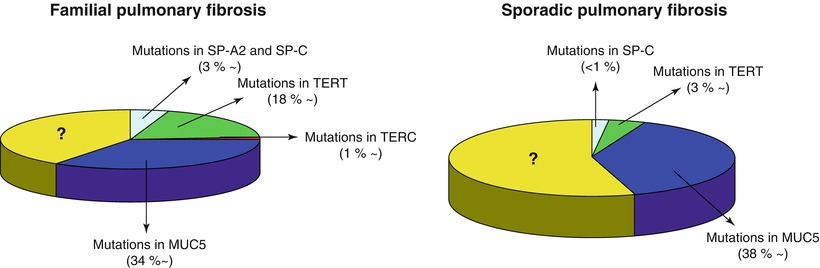
Fig. 36.2
Estimated frequencies of genetic mutations in familial and sporadic cases of pulmonary fibrosis
Hermansky-Pudlak Syndrome
Hermansky-Pudlak syndrome (HPS) is a rare autosomal recessive disorder most commonly found in persons from Puerto Rico characterized by oculo-cutaneous albinism, bleeding diathesis, and accumulation of ceroid, a chromolipid related to lipofuscin, in the reticuloendothelial system of various tissues [10, 11]. Seemingly disparate, these abnormalities are presumably related to defective formation, trafficking, or function of intracellular lysosome-related organelles: melanosomes (oculocutaneous albinism), platelet dense bodies (bleeding dysfunction), and lysosomes (ceroid lipofuscin deposition; [12]). The disease can also be complicated by lung fibrosis, neutropenia and granulomatous colitis.
HPS comprises nine known disorders (HPS-1 to HPS-9), the majority of which present with the same clinical phenotype to varying degrees of severity. Prevalence is estimated at in 1,800 in northwestern Puerto Rico – making it the most common single-gene disorder amongst this population – with only isolated case reports and small case series being reported in the rest of the world [13]. HPS can be caused by mutations in one of several genes: HPS1 (10q23.1), AP3B1 (5q14.1; HPS-2), HPS3 (3q24), HPS4 (22q11.2-q12.2), HPS5 (11p15-p13), HPS6 (10q24.32), DTNBP1 (6p22.3; HPS-7), BLOC1S3 (19q13; HPS-8), PLDN (15q21.1; HPS9). The majority of Puerto Rican patients demonstrate a homozygous 16 base pair duplication in the HPS1 gene, reflecting a founder effect. This genotype represents the most severe of the known mutations and accounts for a high risk of pulmonary disease, hemorrhage, and granulomatous colitis [14]. Once the diagnosis has been confirmed – by means of genotyping, demonstration of absent dense bodies on whole mount electron microscopy of platelets, or accumulation of ceroid in pathological specimens – other family members should be screened for the syndrome and mutations within genes known to cause the disease searched for [15, 16].
Pulmonary fibrosis, the most serious complication, usually presents in the third or fourth decade, appears to be more common in women and affects up to 80 % of all cases with HPS subtypes 1 and 4 [17]. Dysfunction of lamellar bodies in type II pneumocytes, which synthesize, store, and release surfactant, is probably a contributing factor by causing deposition of ceroid and degeneration and death of type II cells [18]. In addition, the early development of pulmonary fibrosis in HPS suggests that environmental or external insults, acting either alone or coupled with abnormal repair mechanisms, play a major role in producing this complication. Chest radiograph and high resolution computed tomography (HRCT) show nonspecific patterns of diffuse, peripheral reticulation, subpleural cysts, and ground glass opacities. As the disease progresses, peribronchovascular thickening and bronchiectasis develop [19]. Honeycombing, when presents, prevail in the lower lobes but is not predominantly subpleural as in usual interstitial pneumonia (UIP). Similarly, the active fibroblastic foci seen in UIP are generally not present in HPS [20]. HRCT findings tend to differ somehow as the disease progresses. In cases of minimal severity, the most common abnormalities include interlobular septal thickening, reticular opacities, and ground glass attenuation, while in severe cases, subpleural honeycombing and bronchiectasis become the predominant features [19]. Histologic examination, in addition to the extensive fibrosis of the alveolar walls, reveals macrophages filled with ceroid throughout the interstitium and alveolar spaces. Lung disease is the most frequent cause of death, followed by inflammatory bowel disease and bleeding complications. There is no effective treatment for HPS-related pulmonary fibrosis. Although one trial reported delayed disease progression with the anti-fibrotic agent pirfenidone in HPS type 1 patients with forced vital capacity >50 % [21], lung transplantation remains the only potentially life-extending therapy for progressive lung disease.
Telomerase-Associated Pulmonary Fibrosis
Telomeres – the tandem repeats of TTAGGG – represent a molecular cap of non coding DNA that protects the ends of the chromosomes against degradation. With repeated cell division telomeres tend to shorten and the chromosomes may become unstable, fused, or lost, leading to cell apoptosis. A complex of proteins and RNA called telomerase is essential in extending telomeres, thus preventing their shortening: the reverse transcriptase component TERT and the RNA template component TERC are key components of the telomerase complex [22]. In addition to age and telomerase function, environmental factors, such as smoking, affect telomere function by reducing their length [23].
Dyskeratosis Congenita
Dyskeratosis congenita (DC) is a rare systemic disease (overall incidence 1 in 1,000,000 persons) usually presenting in the first or second decade with bone marrow failure, the leading cause of death, and a triad of muco-cutaneous lesions including patchy skin hyperpigmentation of the upper chest and neck, dystrophic nails, and oral leukoplakia [24]. DC is considered a syndrome of premature aging as suggested by other common features, such as premature graying of the hair, pulmonary fibrosis, testicular atrophy, cryptogenic cirrhosis, osteoporosis, and increased risk of malignancy. Further corroborating this hypothesis, DC has been the first disease recognized to result from impaired telomere maintenance [25]. Mode of inheritance may be autosomal dominant, recessive, or X-linked, with this latter form resulting from a mutation in the DKC1 gene, which encodes dyskerin, a telomerase-associated protein. Similarly, there is wide variation in severity and spectrum of manifestations, which is only partially explained by locus and allelic heterogeneity. Affected cases with mutations in the genes for TERT and TERC often demonstrate genetic anticipation, the occurrence of more severe and earlier onset disease in later generations secondary to the progressive shortening of telomeres [26]. While telomere length is uniformly reduced, thus indicating a shared mechanism, a genetic defect has not yet identified in most patients with DC, an indication of the complexity of the pathways involved in this disease.
Pulmonary fibrosis develops in approximately 20 % of cases. The histology most often demonstrates UIP, although other patterns including bronchiolocentric inflammation and nonspecific interstitial fibrosis have been described. Pulmonary complications often arise after stem cell transplantation used to treat bone marrow failure [25]. Prognosis of DC patients after the development pulmonary fibrosis is poor, with death occurring 12–40 months after the onset of dyspnea [27].
Short telomere length occurs also in familial and sporadic cases of pulmonary fibrosis without other features of DC. In a cohort of subjects with familial interstitial pneumonia 6 of 73 probands (8 %) had heterozygous mutations in TERT or TERC in circulating lymphocytes [5]. Asymptomatic subjects with mutant telomerase also had short telomeres, suggesting that they may be at risk for the disease. In a larger cohort with both familial and sporadic pulmonary fibrosis, the lymphocyte telomere length was uniformly less than 10 % of age-matched controls in cases with identifiable telomerase-associated mutations. Similarly short telomeres have been found in patients without identifiable mutations. The overall incidence of short telomeres was 25 % of sporadic and 37 % of familial cases of pulmonary fibrosis [4]. Reduced telomere length has also been reported in both alveolar epithelial cells and lymphocytes in sporadic cases of idiopathic interstitial pneumonia – with patients displaying significantly shorter telomeres compared to age-matched controls – even when no telomerase mutations are identified [28]. Furthermore, there is evidence that short telomere length may be a risk factor for disease outside the lung, such as liver cirrhosis or diabetes [28].
Lysosomal Lipid Storage Disease
Lysosomal lipid storage diseases, also known as lip doses, are inherited metabolic disorders in which lipids accumulate in cells and tissues. They represent about 70 genetically distinct conditions with a combined birth frequency of about 1 in 7,500 [29]. Complex lipids, such as glycosphingolipids, which have a major structural function in many cell types, are constitutively degraded within the endolysosomal system by soluble hydrolytic enzymes with the help of lipid binding proteins in a sequential manner. Due to a functionally impaired hydrolase or auxiliary protein, their lipid substrates cannot be degraded, accumulate in the lysosome and slowly spread to other intracellular membranes. In most lysosomal lipid storage diseases the accumulation of one or few lipids leads to the co-precipitation of other hydrophobic substances in the endolysosomal system, such as lipids and proteins, causing a “traffic jam”, thus impairing lysosomal function, such as delivery of nutrients through the endolysosomal system and leading to a state of cellular starvation.
Gaucher’s Disease
Gaucher’s disease – the most common lysosomal lipid storage diseases – is an autosomal recessive disorder caused by deficient lysosomal β-glucosylceramidase activity with subsequent tissue deposition of glucosylceramide [30]. Gaucher’s disease has been identified throughout the world and among all ethnic groups. The pathologic hallmark is the presence of the so-called Gaucher cells in the macrophage-monocyte system, particularly in the liver, spleen and bone marrow. These cells, which are 20–100 μm in diameter, have a characteristic wrinkled-paper appearance resulting from intracytoplasmic substrate deposition, and stain positively with PAS. Gaucher’s disease is classified into three broad phenotypes based upon the presence or absence of neurological involvement: Type 1 (non-neuronopathic), Type 2 (acute neuronopathic), and Type 3 (subacute neuronopathic) [30].
The β-glucosylceramidase gene is located to 1q21 and comprises 11 exons and 10 introns, spanning 7.6-kb of sequence. Nearly 300 mutations within the β-glucosylceramidase gene have been identified in Gaucher patients – including frame-shift mutations, point mutations, deletions, insertions, splice site mutations, and recombinant alleles [31] – with a distribution that spans the gene. Based on the level of glucosylceramidase production, these mutations are commonly classified as null, severe or mild. In the presence of null mutations, such as c.84dupG (84 GG), there is no enzyme production, while severe mutations, such as c.1448 T > C (L444P), though leading to enzyme production, are usually associated with Type 2 or 3 disease when inherited with a null or another severe mutation. On the other hand, mild mutations, such as c.1226A > G (N370S), are only associated with Type 1 disease [32]. Gaucher patients display a wide spectrum of clinical phenotypes, ranging from asymptomatic adults to children who succumb from devastating neurological disease. Hepatosplenomegaly, haematological abnormalities and orthopaedic complications represent the predominant manifestations [33]. Conversely, pulmonary involvement clinically manifests in less than 5 % of patients with Type 1 disease [34]. Four pattern of pulmonary involvement have been described: intracapillary, patchy interstitial infiltrates in a lymphatic distribution, massive interstitial thickening of alveolar septa, and intra-alveolar infiltrates. Respiratory manifestations, which include recurrent infections leading to progressive dyspnea (often culminating in fatal respiratory failure), result from infiltration of alveolar, interstitial, perivascular, and peribronchial spaces by Gaucher cells. Accordingly, chest X-ray and HRCT show bilateral interstitial infiltration, in the form of either a predominant ground glass pattern with superimposed thickening of interlobular septa or a diffuse reticulonodular infiltrate [35]. L444P homozygotes appear at major risk for developing pulmonary disease, even at an early age [35]. On the other hand, pulmonary hypertension, strongly associated with splenectomy and female gender, may occur in subjects with non-N370S mutation within β-glucosylceramidase gene, positive family history, and angiotensin converting enzyme I gene polymorphism [36]. Despite significant advances in our knowledge of the spectrum of mutations within β-glucosylceramidase gene, our ability to make prognostic predictions from genotypic data remains limited. While it is possible to enumerate individual mutant alleles encountered in patients with Gaucher’s disease Type 1, 2, and 3, it is the combination of mutations on both alleles that is important in defining the phenotype. In addition, similar phenotypes may result from different genotypes, while individuals sharing the same genotype can present with and exhibit different disease manifestations, clinical courses and responses to therapy [31]. Gaucher disease is the first lysosomal lipid storage diseases to be successfully treated by enzyme replacement therapy [37]. At present, alglucerase (Ceredase®, Genzyme Inc.), imiglucerase (Cerezyme®, Genzyme Inc.), and velaglucerase alfa (VPRIV™, Shire) have been FDA-approved for treatment of Gaucher patients. However, enzyme replacement therapy, which consists of intravenous infusions of recombinantly produced glucocerebrosidase, is a costly and lifelong treatment. In addition, in contrast to the remarkable effect on hepatosplenomegaly and haematological abnormalities, pulmonary manifestations (clinically, functionally, and radiologically) appear to respond only poorly to enzyme replacement therapy [38]. Similarly, because it does not cross the blood-brain barrier, enzyme replacement therapy does not prevent or halt neurologic involvement.
Niemann-Pick Type Disease
The eponym “Niemann-Pick disease” (NPD) is commonly used to designate a heterogeneous group of autosomal recessive lysosomal lipid storage diseases that share the general clinical and biochemical features of hepatosplenomegaly, with varying degrees of sphingomyelin and cholesterol accumulation in reticuloendothelial and parenchymal tissues, with or without neurological involvement [39]. NPD is commonly classified in three major subgroups: type A is characterized by severe, early central nervous system deterioration and massive visceral and cerebral sphingomyelin storage resulting in death in the first few years of life; type B has a chronic course with marked visceral involvement but a sparing of the nervous system; type C is characterized by a subacute nervous system involvement with a slower course and a milder visceral storage. NPD types A and B are caused by mutations in the sphingomyelin phosphodiesterase 1 gene (11p15.1–11p15.4) that result in deficient lysosomal acid sphingomyelinase activity. Of the mutations in the sphingomyelin phosphodiesterase 1 gene causing types A and B NPD only a few occur frequently. Instead, most of them are “private,” having been described only in one or a few families. Frameshift mutations, small and large insertions and deletions and splicing defects typically have little or no residual acid sphingomyelinase activity and are called type A alleles. Conversely, mutations that retain significant residual activity (>5 % of in vitro-expressed wild-type activity) are neuroprotective and are called type B alleles [40]. Inheritance of two type A alleles predicts a type A phenotype with a neurodegenerative disease course. In contrast, inheritance of one type B allele is neuroprotective and predictive for a type B phenotype, even if the other allele has a type A abnormality. Niemann-Pick C (NPC) disease is a neurovisceral lysosomal lipid storage diseases characterized by abnormal intracellular transport of endocytosed cholesterol with sequestration of unesterified cholesterol in lysosomes and late endosomes [41]. The disease is transmitted in an autosomal recessive manner and is caused by mutations in either NPC1 (18q11-q12, 95 % of cases) or NPC2 (14q24.3) genes, with nonsense or frameshift mutations within NPC1 being associated with the most severe neurological course. Although the precise functions of the NPC1 and NPC2 proteins are still elusive, they are thought to function in a coordinate fashion in the cellular post-lysosomal/late endosomal transport of cholesterol and other molecules [42, 43]. The clinical presentation of NPC disease is extremely heterogeneous, with an age of onset ranging from the perinatal period until well into adult age. Likewise, the lifespan of the patients varies between a few days until over 60 years of age, although the majority of cases die between 10 and 25 years of age. Apart from a small subset of patients who die at birth or in the first 6 months of life from hepatic or respiratory failure, all patients will ultimately develop a progressive and fatal neurological disease, which manifests mainly with cerebellar ataxia, dysarthria, dysphagia, and progressive dementia. The majority of cases show also a characteristic vertical supranuclear gaze palsy. Systemic disease – in the form of liver, spleen and lung involvement – always precedes the onset of neurological symptoms [44].
Pulmonary disease most often presents with features of endogenous lipoid pneumonia – with the typical foamy-appearing macrophages containing sphingomyelin (Figs. 36.3 and 36.4) – or, interstitial fibrosis, which produce either a miliary or a reticulo-nodular appearance on the chest radiograph [45, 46]. Niemann-Pick cells (sea-blue histiocytes) are seen in bronchoalveolar lavage fluid [47]. One series of 53 patients with type B disease found evidence of interstitial abnormalities on HRCT in 98 % of cases: upper lobe predominant ground glass opacities and basilar predominant interlobular septal thickening were the most common features (Figs. 36.5 and 36.6) [45]. Pulmonary function test, which commonly shows a mild restrictive defect with minimal alteration of the diffusing capacity for carbon monoxide, do not appear to correlate with diseased extension on HRCT [45]. Improvement of endogenous lipid pneumonia has been reported with whole lung lavage [46]. A first product, miglustat, has been granted marketing authorization in Europe and several other countries for specific treatment of the neurological manifestations.
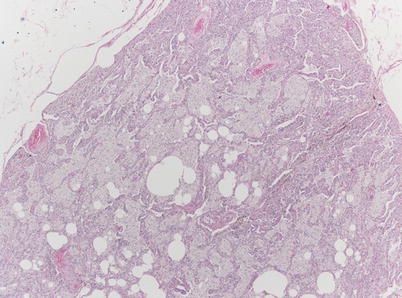
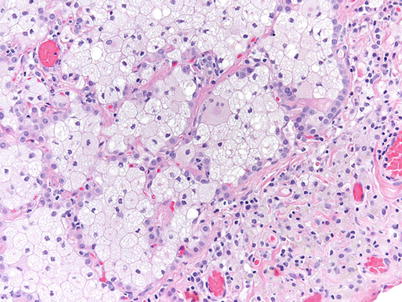
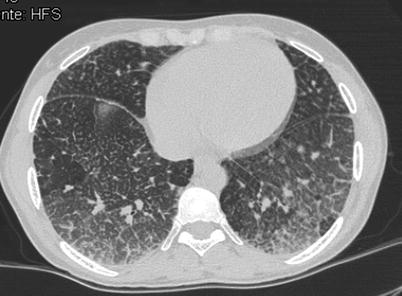
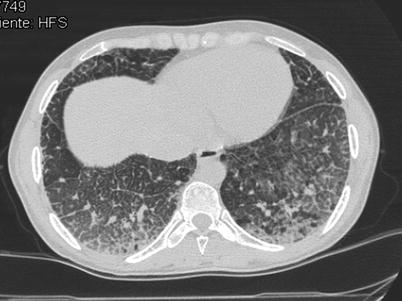
Fig. 36.3
Surgical lung biopsy in a 33-year-old woman with type-B Niemann-Pick disease. At low magnification, alveoli are filled with pale-staining macrophages (hematoxylin-eosin, 20×) (Slide courtesy Alberto Cavazza, MD)
Fig. 36.4
Higher magnification showing the finely vacuolated cytoplasm of the intra-alveolar macrophages (hematoxylin-eosin, 200×) (Slide courtesy Alberto Cavazza, MD)
Fig. 36.5
Type B Niemann-Pick disease. Transverse CT scan of midlung zones in a 33-year-old woman shows severe interstitial changes. Note the presence of ground-glass opacities and the intermixed thickened interlobular septa and intralobular lines in some areas; these findings are suggestive of the “crazy paving” sign
Fig. 36.6
Same patient as in figure 36.5. CT scan shows dense ground-glass opacities and thickened interlobular and intralobular septa “crazy paving”
Fabry’s Disease
Fabry’s disease is an X-linked inborn error of glycosphingolipid catabolism, resulting from a deficient lysosomal α-galactosidase A activity, and characterized by an abnormal accumulation of the glycosphingolipid ceramide trihexoside in vascular smooth muscle throughout the body, particularly in vessels of the skin, kidneys, heart, pulmonary vascular and neurological system [48]. It begins in childhood with a median age of survival of 55 years, although adult patients with less pronounced disease can survive to older ages and be discovered because of cardiac involvement causing a cardiomyopathy [49]. Pulmonary involvement – in the form of diffuse alveolar hemorrhage associated with renal failure (pulmonary renal syndrome) – has occasionally been reported [50]. Conversely, airway obstruction manifesting with dyspnea, cough and wheezing and caused by deposition of glycosphingolipid in the cells lining small airways is a common finding in Fabry’s disease [51]. Previously a universally fatal disease, the recent development of human recombinant α-galactosidase A has been shown to reverse the clinical manifestations of the disease [52].
Lysinuric Protein Intolerance
Lysinuric protein intolerance is an autosomal recessive disease characterized by defective transport of lysine, arginine, and ornithine with excessive loss of proteins in the urine [53]. Lysinuric protein intolerance is caused by mutations in the solute carrier family 7A member 7 (SLC7A7) gene on 14q11.2, which encodes y+ LAT-1 protein, the catalytic light chain subunit of a complex belonging to the heterodimeric amino acid transporter family. Mutations within SLC7A7 – mainly in the form of single-base substitutions or small deletions – have been identified in all but one individuals with lysinuric protein intolerance [54]. Initially described in Finland, lysinuric protein intolerance is a rare disease with fewer than 100 cases reported. Affected individuals demonstrate failure to thrive, growth retardation, hepatosplenomegaly, hypertonicity, and osteoporosis. Pulmonary involvement ranges from asymptomatic interstitial abnormalities to acute and life-threatening acute respiratory failure due to either alveolar hemorrhage or alveolar proteinosis – the latter being the most commonly observed interstitial lung disease [55]. Typically, in alveolar proteinosis the alveolar spaces are invaded by foamy macrophages filled with proteinaceous material, with chest radiograph revealing diffuse alveolar infiltrates. HRCT shows a characteristic pattern of ground glass opacities superimposed over a pattern of fine overlapping lines forming irregular polygonal shapes (“crazy paving” pattern). Similar to other forms of alveolar proteinosis, the treatment of choice is whole-lung lavage [56].
Familial Hypocalciuric Hypercalcemia
Familial hypocalciuric hypercalcemia is a rare autosomal dominant disorder with variable penetrance, characterized by familial hypercalcemia with hypocalciuria, granulocyte dysfunction, and interstitial lung disease (ILD) [57]. Familial hypocalciuric hypercalcemia is caused by inactivating mutations in the calcium sensing receptor gene leading to a calcium-hyposensitivity, compensatory hypercalcaemia – in order to obtain intracellular response despite inactive receptors – and hypocalciuria. The low urine calcium distinguishes familial hypocalciuric hypercalcemia from primary hyperparathyroidism, in which urine calcium excretion is increased. Lung involvement consists of a granulomatous disease, with foreign body giant cells and mononuclear cells infiltrating the alveolar interstitium. There are, however, no well-formed, circumscribed granulomas as observed in sarcoidosis or berylliosis. In addition, contrary to sarcoidosis and berylliosis, urine calcium is normal or low, and the level of 1,25-dihydroxyvitamin D3 is within normal limits. In general, familial hypocalciuric hypercalcemia is a benign condition that does not require treatment. As such, the main argument for establishing the diagnosis is to avoid unnecessary and futile parathyroidectomy. However, in chronic severe cases, lung fibrosis and honeycomb changes may lead to respiratory failure, thus reducing life expectancy. Chondrocalcinosis and acute pancreatitis have also been reported [58, 59]. The third feature of familial hypocalciuric hypercalcemia is granulocyte dysfunction due to a myeloperoxidase deficiency and reduced antistaphylococcal killing [60].
Neurofibromatosis Type 1
Neurofibromatosis 1 (NF1), previously known as von Recklinghausen’s disease, is a systemic disorder affecting approximately 1 of 3,500 persons worldwide and caused by loss-of-function in the NF1 gene (17q11.2) that encodes the tumor suppressor protein neurofibromin [61]. Despite its inheritance pattern – autosomal dominant – as many as 50 % of cases are spontaneous, that is caused by a de novo NF1 mutation. NF1 is recognized by the appearance of multiple (>6) café-au-lait spots, axillary and inguinal freckling, optic gliomas, osseous lesions and cutaneous neurofibromas; internal neurofibromatous tumors are most commonly found in the central nervous system [62]. In addition, Lisch nodules, pigmented hamartomatous nodular aggregates of dendritic melanocytes affecting the iris, are present in over 90 % of patients. The spectrum of clinical phenotypes and their development, severity, and prognosis is large and is thought to result from the cross talk between numerous cell types, cell signaling networks, and cell-extracellular matrix interactions. At one end of this spectrum the lifetime risk of malignant tumors arising from peripheral nerves, which is estimated to be 10–13 % [63].
Interstitial lung disease, which usually appears between the ages of 35 and 60 years, complicates 6–12 % of cases [64]. The chest X-ray typically demonstrates – alone or in combination – lower zone (usually symmetrical) reticulo-nodular infiltrates and bullous changes (usually asymmetrical) in the upper zones [65]. HRCT reveals bibasilar reticular changes, ground glass opacities, upper lobe predominant small cysts and bullous changes [66]. Thin-walled bullae are present in almost all patients with ILD, although they may be seen in isolation. Histologically, alveolar septal fibrosis represents the major change, while an alveolitic process consisting of a mononuclear cell infiltration may be observed in earlier disease [67]. Functionally, NF1-associated ILD is characterized by the gradual appearance and slow progression of a mixed obstructive and restrictive ventilatory defect. NF1-associated ILD is often progressive and may lead to pulmonary hypertension and right heart failure [68]. Rarer pulmonary complications of NF1 include large-airway obstruction, mediastinal, bronchial or intraparenchymal neurofibromas (leading to diaphragmatic paralysis), scar carcinoma complicating fibrotic lung disease, primary lung cancer developing in the walls of emphysematous cysts, and pneumothorax [69–71]. The pathogenesis of the ILD in NF1 is unknown and no effective therapies are currently available.
Surfactant Dysfunction Disorders
Surfactant dysfunction disorders are caused by mutations within genes encoding proteins needed for normal function and metabolism of surfactant, a mixture of phospholipids and proteins synthesized, packaged, and secreted by alveolar type II cells that lowers surface tension and prevents atelectasis at end-expiration. While rare, these disorders are associated with considerable pulmonary morbidity and mortality.
Surfactant Protein B (SFTPB) Deficiency
Surfactant protein B (SFTPB) deficiency is a rare autosomal recessive disease responsible for rapidly progressive neonatal respiratory distress syndrome [72]. Over 40 loss-of-function mutations within SFTPB gene that result in partial to complete absence of SP-B protein have been identified thus far. The most common one – a GAA substitution for C at genomic position 1,549 in codon 121 (the “121ins2” mutation), which accounts for approximately 70 % of cases of SFTPB deficiency – results in an unstable transcript and consequent absence of pro- and mature SFTPB protein [73]. The absence of SFTPB, in turn, leads to abnormal surfactant composition, decreased surfactant function, and structural disruption of lamellar bodies. Accordingly, SFTPB deficiency is characterized histologically by the accumulation of granular, eosinophilic, periodic acid-Schiff-positive, lipoproteinaceous material in the alveolar spaces, which often contains desquamated alveolar type II cells and foamy alveolar macrophages.
Clinical estimates suggest an incidence of 1 per million live births. Most infants with SFTPB deficiency present within hours of birth with respiratory failure requiring mechanical ventilation. Chest radiograph and HRCT appearance mimics that of hyaline membrane disease in premature infants with diffuse haziness and air bronchograms. However, infants with SFTPB deficiency are only transiently or minimally responsive to surfactant replacement and, with rare exceptions, all patients succumb without lung transplantation. Children with mutations that allow for partial expression of the SFTPB protein appear to survive longer and go on to develop a chronic ILD [74].
Surfactant Protein C (SFTPC) Deficiency
Surfactant protein C (SFTPC) deficiency is a rare disorder originally described in an infant with nonspecific interstitial pneumonia (NSIP) whose mother had desquamative interstitial pneumonia. Both carried an heterozygous guanine to adenine substitution leading to skipping of exon 4 and deletion of 37 amino acids [2]. A large five generation kindred was later described with 14 affected family members, including four adults with surgical lung biopsy evidence of UIP and three children with NSIP, all carrying a rare heterozygous missense mutation substituting a polar residue (glutamine) for a neutral one (leucine) predicted to hinder processing of SP-C precursor protein [1]. Over 35 dominantly expressed mutations within SFTPC gene have been identified, half of which arise spontaneously, thus resulting in sporadic disease, whereas the remaining are inherited. The most common mutation, a T to C transition at genomic position 1,295, results in a threonine substitution for isoleucine in codon 73 (I73T), and accounts for a quarter of cases [75].
The pathophysiology of lung disease due to SFTPC mutations is complex and is thought to be related to aberrant surfactant protein folding, decreased endogenous SP-C secretion, endoplasmic reticulum stress and apoptosis of alveolar type II cells [76]. The age of onset and severity of disease are highly variable, ranging from fatal respiratory distress in infants to pulmonary fibrosis in older adults. In a recent study from the Netherlands SFTPC mutations accounted for as many as 25 % of familial pulmonary fibrosis kindreds [77]. Conversely, mutations within SFTPC are rarely associated with sporadic forms of pulmonary fibrosis [78]. Whether the nature and location of SFTPC mutations impact on severity of lung disease is unknown. However, affected family members harboring the same SFTPC mutation display considerable variability in the onset and severity of lung disease [1]. The variability in the natural history of the disease precludes accurate assessment of prognosis for the individual patient and complicates interpretation of potential drug therapies.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


