DAMAGE VERSUS MORBIDITY
The terms damage and morbidity are often used interchangeably, but they are quite different concepts. In clinical practice, some degree of radiation-induced lung damage, either pneumonitis or fibrosis, is considered acceptable because of the large functional reserve of this tissue. The whole organ does not fail to function if some part of it is destroyed. Morbidity, on the other hand, is a clinical term that describes how well an individual patient feels or how well a specific organ functions. The morbidity of treatment is determined by many factors, including the damage to the tissue, the effect of this damage on organ function, and the effect of both of these factors on the patient’s well-being and lifestyle. Because of the functional reserve of the lung, structural damage is not necessarily reflected in clinical morbidity, assessed in terms of whole-lung function.
The dissociation between damage and morbidity in the lung reflects the organization of those anatomical units responsible for lung function.
3 Morbidity is a reflection of two parameters: (a) the survival of sufficient numbers of cells to maintain tissue function and (b) the organization of those cells into units that carry out tissue function. The spatial relationship between these functional subunits differs between tissues and is a critical determinant of the relationship between damage and any resultant morbidity.
Anatomically, the lung is a system of branching ducts and accompanying blood vessels that ultimately terminate in the alveoli, the site of gas exchange. The functional subunit in the lung is most likely the
acinus, which is structurally well defined, beginning at the ramification of the terminal bronchiole to the respiratory bronchioles and terminating in the alveolar sacs, each of which bears numerous alveoli. Each acinus is a self-contained entity independent of its neighbor. Presumably, destruction of one acinus will have no measurable effect on lung function in a normal healthy lung, and functional damage, particularly total lung function, will be manifested only
when a critical number of these units are destroyed. A useful analogy is a strand of sequential lights arranged in parallel. When one burns out (damage), none of the others is affected, and the overall effect is not diminished; that is, the damage is below the threshold of the human eye to distinguish it (no morbidity). However, when sufficient numbers of lights burn out such that damage is noticeable (the threshold for damage detectable by the human eye is exceeded), the overall effect is diminished (morbidity occurs).
Many assays are available to quantify radiation-induced damage and morbidity in the lungs in experimental animals and in patients. In humans, regional pulmonary function or the extent of pneumonitis and fibrosis evident on radiographs can be quantified by using functional imaging modalities such as single photon emission computed tomography (SPECT),
18 F-fluorodeoxyglucose positron emission tomography (FDG-PET), and ventilation planning computed tomography (CT), thereby providing systems by which damage can be scored.
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15 In rats and mice, the experimental animals most commonly used to study radiation-and drug-induced lung injury, measurements of regional lung function are difficult and pulmonary function tests most often assess total lung function, a measure of morbidity.
16,
17,
18,
19,
20 The diffusion capacity, measured as DLCO (diffusing capacity of lung for carbon monoxide) is the most useful clinical tool of functional lung oxygen exchange. When sufficient functional reserve units are destroyed, either as preexisting lung damage from smoking, or as new damage from treatment, this can be a sensitive measure of that injury. In patients, structural damage can be quantified by noninvasive methods, for example by CT scanning; in experimental animals, such damage is most often assessed in autopsy specimens. In this chapter, clear distinctions will be made between damage and morbidity and the assessment of each.
RADIATION-INDUCED LUNG DAMAGE
Pathophysiology Radiation-induced damage to the lung occurs in distinct phases characterized by differences in when after irradiation they occur and their histologic or molecular manifestations (
Table 44.1).
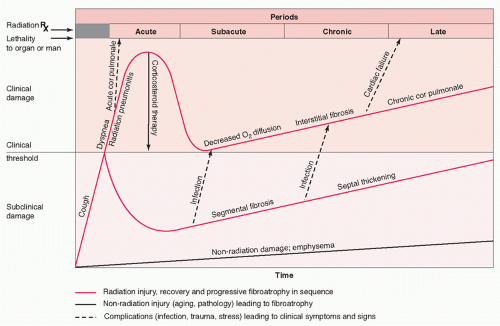
21 Damage to the human lung has long been described as occurring in four clinical phases: a phase
of acute injury termed
radiation pneumonitis, a subacute phase, a chronic phase characterized by lung fibrosis, and a late phase (
Fig. 44.1).
22,
23,
24,
25,
26 Two of these phases are clearly separated in time: pneumonitis occurs from 3 to 6 months after the beginning of treatment, whereas fibrosis occurs from 1 year onward. Both pneumonitis and fibrosis have been well defined histopathologically under controlled conditions in animals. However, because animals can be sacrificed and studied at predetermined times after irradiation, an additional earlier, asymptomatic phase of damage has been defined—the latent phase.
26,
27,
28 The characteristics of radiation-induced lung damage during each of these five phases are described further in the following sections.
Latent Phase The weeks to months preceding the overt appearance of radiation pneumonitis is referred to as the “latent” period, because no overt histopathologic, radiographic, or clinical signs and symptoms of radiation damage can be observed. In most cases, overt pulmonary reactions are not expressed either clinically or histologically in humans or in animal models for the first 2 or 3 months after irradiation, regardless of the volume of lung irradiated. Although no changes can be observed at the light microscopy level during this latent period, electron microscopy reveals degranulation and loss of type II cells (with attendant loss of surfactant), loss of basal laminar proteoglycans (resulting in swelling of the basement membrane), and transudation of proteins into the alveolar spaces (indicating increased capillary permeability and suggesting a loss of endothelial cells) within the first month after whole-lung irradiation.
24,
25,
27 Endothelial cells themselves become vacuolated and pleomorphic and may slough, leading to denudation of the basement membrane and changes in capillary permeability.
29 Although these changes are dose related and diffuse throughout the lung, they are not in themselves sufficiently severe to result in death during this time. Deaths do not occur before overt histologic damage appears.
It was formerly assumed that during the latent phase, a series of biochemical events occurred later manifest as overt
expression of damage during the next phase of lung injury (pneumonitis), although no such events had been identified. New molecular techniques and tools suggest that dramatic changes do occur during this period and, depending on the radiation dose, may resolve or may progress to the next phase, which is characterized by overt signs and symptoms of radiation pneumonitis. These molecular changes are discussed next.
Acute Phase: Classic Radiation Pneumonitis Evidence of structural changes in the lung appear within the first 6 months after irradiation of all or part of the lung of humans or experimental animals, resulting in diffuse alveolar damage.
1,
26,
27,
28 Although this phase of damage occurs relatively long after the lung irradiation, histologically, it is an acute effect that is characterized by a prominent inflammatory cell infiltrate consisting of macrophages, lymphocytes, and mononuclear cells in the pulmonary interstitium, which is normally devoid of cells, and in the air sacs.
3,
22,
24,
25,
26,
27,
28 In animal models, neutrophils usually do not predominate in this inflammatory cell infiltrate. Although loosening and widening of the interstitium indicates interstitial edema, only after high doses is edema observed in the air spaces. When the whole of both lungs is irradiated in humans or experimental animals, the damage is diffuse and, if sufficiently severe, fatal.
This acute phase of damage in the lung is generally referred to as
pneumonitis, a term that usually refers to an inflammatory reaction in the lung caused by local growth of bacteria, fungi, or parasites. In such situations, the cellular infiltrate contains polymorphonuclear leukocytes, a cell type rarely found in the “sterile” inflammatory infiltrate in the irradiated lungs of experimental animals. When present, these cells are indicative of a superimposed infection and the cause of death—radiation, infection, or both—is unclear. Perhaps a more appropriate term for this phase of diffuse alveolar damage after lung irradiation is
alveolitis, which refers to an inflammatory reaction, in this case a pathological condition not caused by a microorganism.
30 Although an inflammatory cell infiltrate in the interstitium and the air sacs is a prominent component of radiation alveolitis, the relative contribution of these inflammatory events versus direct tissue injury from radiation in the pathogenesis of this syndrome is unclear.
In mice, pneumonitis begins at 3 months after radiation is administered and persists for up to 6 months, with most deaths occurring between 4 and 5 months posttreatment. The latency period for the appearance of damage depends on the radiation dose, appearing sooner after high doses than after low doses. To account for the dose-dependent difference in the latency period and to include all responders in the assay, researchers set the standard time for scoring deaths from pneumonitis in animal experiments between 3 and 7 months. Techniques used to quantify this phase of lung damage in experimental animals include functional assays such as breathing rate
16,
18,
20 and carbon monoxide uptake
17; CT scans
19; quantitative morphometry
28,
31; and, of course, lethality from the syndrome.
3 These measures provide steep dose-response curves from which estimates of an effect dose for a given severity of injury can be obtained. Generally, the dose that prompts a certain effect in 50% of animals (ED
50) or the dose that kills 50% of the population (LD
50) is used. In mice, estimates of the LD
50 from radiation pneumonitis occurring between 3 and 6 months after whole-lung irradiation range from 9 Gy to greater than 16 Gy, depending on the mouse strain. In the clinic, the use of large single doses to the upper half of the body or to the whole body has provided information on radiation dose-response curves for radiation pneumonitis in humans.
32 When either the incidence of pneumonitis
33,
34,
35,
36 or evidence of damage on CT scans
9,
37 is used to quantify increases in lung density in patients, the shape of the resultant dose-response curves for lung damage in humans parallels those for mice. The ED
50 for pneumonitis in humans, 9 to 10 Gy, is within the range of LD
50 s for pneumonitis measured for different mouse strains, although on the low end of the range. In one study, the relationship between regional dose and radiation pneumonitis response was linear when pneumonitis was evaluated with FDG-PET/CT imaging.
14 Although treatment of lung cancer can involve irradiation of large volumes of lung, rarely is the whole lung irradiated, and thus, fatal pneumonitis is relatively uncommon, occurring in 1% to 4% of cases.
38,
39 However, that the range in irradiated patients is 2% to 33% for severe pneumonitis,
38,
39,
40,
41,
42 and an even higher proportion (up to 77%) demonstrate CT scans lung changes consistent with pneumonitis.
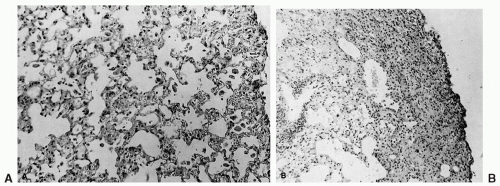
Previously, diffuse alveolitis had been considered characteristic of the acute phase of radiation-induced lung damage in humans and animals, but the histopathologic differences between mouse pneumonitis and human pneumonitis could not be explained. For example, a characteristic histologic finding in irradiated human lung is the presence of hyaline membranes. Mice do not develop fibrosis during the pneumonitis phase, but focal areas of fibrosis have been reported in patients within the first month or two after lung irradiation, the time generally associated with the infiltrative, exudative lesions of radiation pneumonitis. These discrepancies between mice and humans have been partially resolved by the work of Sharplin and Franko,
43 who report that the pathology depends on the strain of mouse used. For example, mice of the C3H and CBA strains showed a classic diffuse alveolitis (pneumonitis) without evidence of fibrosis, whereas the C57B16 strain exhibited protein-rich edema, hyaline membranes, and focal fibrosis, with few of the changes characteristic of pneumonitis (
Fig. 44.2). These findings indicate that the choice of mouse strain is critical in studies of radiation lung damage. In fact, mouse strains used to study pneumonitis would be different from those chosen to study fibrosis. Clearly, the mechanism of damage varies between strains, and the underlying mechanisms may vary in humans as well.
Intermediate (Subacute) Phase If the radiation dose is low, or if less than a critical volume of lung is irradiated, the acute pneumonitis phase resolves. Although few data are available on this phase in humans, irradiated mouse lung experiments
provide a model.
18 In mice that survive the acute pneumonitis phase, lung function, as measured by breathing rate, improves. The lungs, though, are not totally normal, and foci of foamy macrophages with hyperplasia of type II epithelial cells in the air spaces are the dominant findings; however, no deaths occur during this phase of damage. Thus, this phase appears to be one of resolution of the early exudative alveolitis.
Late Phase: Radiation Fibrosis In contrast to the effects of acute alveolitis, the effects of radiation in humans that are chronic are observed from months to years later, even though biochemical and histologic changes occur months earlier. Pulmonary fibrosis develops insidiously and may stabilize after 1 to 2 years. Although numerous studies have attempted to elucidate the mechanism(s) leading to pulmonary fibrosis, the pathogenesis of this late lung response remains elusive and controversial.
Pulmonary fibrosis is the end stage of a complex process of abnormal repair of damage that may be preceded by an inflammatory response dominated by macrophages and lymphocytes. Radiation-induced lung fibrosis is generally thought to be the repair process that follows radiation pneumonitis or radiation alveolitis. Fibrosis of the pulmonary parenchyma may occur as a diffuse or focal lesion, but the designation of pulmonary fibrosis is usually reserved for diffuse or widespread multifocal collagen deposition involving the peripheral air spaces. Although often termed
interstitial fibrosis, collagenous thickening of alveolar walls is often a consequence of the incorporation of intra-alveolar exudate into the interstitium and the subsequent reepithelialization, which leads to a revision of alveolar architecture. In fact, it is often suggested that severe exudative alveolitis of long-standing duration results in a generalized fibrotic thickening of the alveolar septa.
30,
44 Loss of pulmonary function results from focal microcollapse of alveoli and apposition of alveolar walls, leading to irreversible remodeling of pulmonary architecture.
Although lung irradiation studies in mice indicate that pneumonitis and fibrosis are directly related,
28,
45,
46,
47 other studies, also in murine models, show that these two sequelae of lung irradiation can be dissociated from each other. They also indicate that radiation-induced lung fibrosis can and does occur without a preceding inflammatory event.
48,
49,
50 These studies suggest that this phase of injury may result from different pathogenic mechanisms than those underlying pneumonitis.
In mice, the exact form of lung fibrosis that occurs after lung irradiation depends on the strain. In early studies of radiation-induced lung damage, mice were killed at 7 months after irradiation, when the pneumonitis phase of damage ended. However, in later studies, mice surviving to 7 months were followed for periods of up to a year after thoracic irradiation. A diffuse thickening of the alveolar septa characterized by collagen deposition was observed at necropsy in these longsurviving mice.
49,
50 The air spaces were clear and patent, and pulmonary architecture was preserved, though the lungs were stiff. Sharplin and Franko
31,
43 reported that radiation-induced lung fibrosis in mice was not always manifested as a diffuse thickening of the alveolar septa and that some strains exhibited atelectasis accompanied by collagen deposition in the collapsed area, resulting in a focal contracted scar. In this form of lung fibrosis, alveolar architecture is obliterated. We too have found a difference in the fibrotic lesions in irradiated mouse lung, with the C57B1/6J mice exhibiting collapsed atelectatic alveoli with superimposed collagen and the C3H strain showing a more diffuse fibrosis of the alveolar walls and small stellate scars surrounded by patent alveoli.
51 In C57B1/6J mice, the collagen appears in organized bundles, making it easily distinguishable on light microscopy with collagen-specific stains; in C3H mice, the initial deposition of the collagen as fine, wispy fibrils in the alveolar wall makes this type of fibrosis more difficult to resolve with light microscopy. In addition to these distinct histologic features, these two forms of fibrosis are
distinguished further by the time at which they appear in the two strains after irradiation. Fibrosis in the C57B1/6J strain occurs within the first 6 months after irradiation, a period during which only pneumonitis is found in the C3H strain. In the C57B1/6J strain, fibrosis of the alveolar walls does not occur until 9 months (or later) after irradiation.
The suggestion from these mouse studies that the pathogenesis of radiation-induced pulmonary fibrosis may not be uniform across strains suggests that there may be three different pathways to lung fibrosis that may depend on the toxic agent:
luminal fibrosis, in which granulation tissue buds into the air spaces;
mural fibrosis, in which exudate is incorporated into the alveolar walls; and
atelectatic induration, which involves partial or complete collapse of alveoli and permanent apposition of alveolar walls followed by fibrous tissue proliferation and collagen deposition in the area.
30 Franko et al.
52 have suggested from breeding studies in inbred strains of mice that radiation-induced lung fibrosis results from two independent pathways that may involve different genes. Further support for the hypothesis that pulmonary fibrosis can arise through several mechanisms comes from studies of the colon, in which two distinct types of fibrosis have been shown to occur after irradiation, possibly as a result of two distinct mechanisms.
53Most studies in mice involve irradiating the whole lung, which is not the standard procedure for radiation-based treatment of lung cancer in humans, in which limited volumes of lung are irradiated. Thus, the question remains as to how relevant findings from experimental animal models are to the radiation-based treatment of lung cancer in humans. For example, hemithoracic irradiation in mice and rats does not produce the same mortality and morbidity as that of whole-lung irradiation because of hypertrophy of the contralateral lung; however, the mechanisms of collapse and fibrosis of the irradiated lung may differ from those that occur after whole-lung irradiation. In studies of the kidney (a paired organ such as the lung), nephrectomy 1 day after bilateral kidney irradiation was found to induce a proliferative response in the remaining irradiated kidney that in turn resulted in partial restoration of kidney function compared with kidney function in irradiated nonnephrectomized mice.
54 Although these differences between animal experiments and radiation-based treatment of humans for lung cancer must be borne in mind, clinical observations indicate that similar-appearing damage occurs in humans and in mice after lung irradiation, supporting the use of these animal models for mechanistic studies. What is most important is that the studies in mice that identified the form and severity of radiation-induced lung fibrosis as being related to the strain of mouse suggest that this late consequence of lung irradiation may be genetically regulated. Identifying, mapping, and cloning the gene(s) for radiation-induced lung fibrosis could provide a means to identify “sensitive” individuals in the population before treatment commences.
Sporadic Radiation Pneumonitis Because one of the characteristic features of classic radiation pneumonitis is that the damage is confined to the irradiated area, it has been suggested that radiation damage that appears outside the irradiated field, anecdotal evidence of which has been available for more than 40 years,
55,
56,
57,
58 represents a hypersensitivity pneumonitis. First suggested by Holt in 1964,
59 this syndrome has been given little attention for two reasons: first, it is rare, and second, in most cases the contralateral lung also received some dose, although the actual quantity was unknown. Three factors suggest that this clinical syndrome may be different from the classic form of radiation-induced lung damage: (a) it affects 10% to 15% of patients; (b) symptoms often resolve without sequelae; and (c) it often develops earlier than classic pneumonitis that is within 2 to 6 weeks after the completion of therapy. In an extensive study of four women treated for breast cancer who experienced bilateral changes after radiation confined solely to one lung, Gibson et al.
60 found marked lymphocytosis in bronchioalveolar lavage samples from both the irradiated and nonirradiated lungs. Gallium scanning confirmed these findings, indicating increased gallium uptake in both the irradiated and nonirradiated lungs. In all four patients, symptomatic improvement was prompt after corticosteroid administration. With these observations, these authors concluded that finding equal abnormalities in both the irradiated and nonirradiated fields was not consistent with simple direct radiation-induced damage but rather implied an immunologically mediated mechanism such as hypersensitivity pneumonitis.
More recently, several investigators have challenged the idea that bilateral radiation pneumonitis after unilateral lung irradiation is relatively uncommon.
61,
62 These authors contend that the extreme dyspnea experienced by patients with radiation-induced pneumonitis seems to be out of proportion to the volume of lung irradiated and cannot be explained on the basis of localized tissue destruction. Calveley et al.
63 investigated the early activation of inflammatory cytokines and macrophages in different regions of the lung after partial volume irradiation and found evidence of an inflammatory response triggered by the partial volume irradiation in the whole rat lung at very early times after irradiation; that response is maintained in a cyclic pattern until later, when the onset of functional symptoms would be expected. The extent of elevation of cytokines and activated macrophages was similar both in the field of treatment and out of it, although more micronuclei were present in field than out of field. Reactive oxygen species induced by this response were thought to play an important role in the induction of both in-field and out-of-field DNA damage.
63 In a prospective study of 17 women with breast cancer, bronchioalveolar lavage analysis and gallium scanning showed diffuse lymphocytosis, increased gallium scan uptake, and decreased alveolar volume and vital capacity in both the irradiated and nonirradiated lungs. Further analysis of the lymphocyte infiltrate in a patient with clinical radiation pneumonitis showed that these cells were almost exclusively recently activated CD4+ helper T cells, cells that have been implicated in hypersensitivity pneumonitis after other insults. In a further analysis of these patients plus an additional
five patients with clinical features of pneumonitis, these investigators found no statistically significant differences between bronchioalveolar findings on the irradiated and nonirradiated sides of the chest either before or after treatment or with or without clinical pneumonitis. Although bilateral lymphocytosis was found in 13 of the 17 patients (76.5%), only 2 had clinical features of radiation-induced pneumonitis. Symptoms resolved spontaneously in the other 11 patients. The other four patients did not demonstrate subclinical or clinical symptoms. Based on these findings, these authors suggested that bilateral involvement of both lungs after the irradiation of one lung represents an immune-mediated hypersensitivity pneumonitis that leads to clinical radiation pneumonitis in only 10% of the irradiated population. Because this syndrome seems to result from an entirely different pathogenetic mechanism than classic radiation pneumonitis, these authors suggested that this form of radiation-induced lung damage be distinguished from the classical form and be called “sporadic radiation pneumonitis.” What distinguishes this type of lung damage from the classic type is that it occurs in an unpredictable manner and involves nonirradiated portions of the lung.
At this time, no animal model for sporadic radiation pneumonitis has been identified. However, in a study of CT changes after irradiation of only the right lung of rats, Geist and Trott
64 observed fluctuations in the radiologic density in the shielded left lung of the irradiated rats that were greater than those observed in control rats and parallel to those occurring in the irradiated right lung, similar to the sporadic pneumonitis in humans. However, a study of rabbits indicated that irradiation of one lung resulted in a decrease in the number of alveolar macrophages, but only in the irradiated lung.
65
Pathogenesis
The Target Cell Concept It has been hypothesized that radiation depletes some critical “target” cells in the lung and that this depletion after a latent period, initiates late sequelae of pneumonitis and fibrosis.
66 The expression of radiation damage also is generally thought to require cell division. Therefore, in tissues like the lung in which damage is not overtly expressed for months after treatment is completed, the long latency period was thought to reflect slowly proliferating target cells. For these reasons, a major research effort has been mounted to identify the target cells with the intention of protecting them from radiation damage, and thereby preventing or at least minimizing the severity of radiation-induced pneumonitis and fibrosis without compromising tumor cell kill and tumor control. The two most likely candidate target cells were thought to be type II cells
26,
67,
68,
69,
70,
71,
72,
73,
74 and vascular endothelial cells.
26,
75,
76,
77,
78,
79,
80,
81,
82,
83,
84 Type II cells divide more frequently than do other types of lung parenchymal cells. They are responsible for synthesizing, storing, and secreting surfactant, the surface-active material that prevents alveolar collapse. Vascular endothelial cells also divide more quickly than do other cell types in the lung. Furthermore, edema, a consistent finding in the interstitium and air spaces after lung irradiation, indicates vascular leakage, which in turn implies the radiation-induced killing and depletion of vascular endothelial cells.
Type II Cells The first evidence to support the hypothesis that type II cells were the target cells for radiation pneumonitis was published in 1982 by Shapiro et al.,
69 who showed that in mice, local irradiation of both lungs with a range of single doses of x-rays produced dose-dependent changes in phospholipids in lung lavage fluid and in lung tissue as early as 24 hours after irradiation. These changes persisted for the first 4 weeks after irradiation, indicating that changes in the function of type II cells occurred long before tissue damage was perceivable with light microscopy. Although the relationship of these early events to the later incidence of pneumonitis and fibrosis was unclear, these findings were the first indication that the latent period was not really latent at all. These investigators then sought to identify biochemical markers of surfactant that could be assessed in patients during radiation therapy, before the onset of clinical and pathologic pneumonitis, with the aim of identifying patients at high risk of developing severe radiation pneumonitis.
85 Their discovery that surfactant apoprotein in the serum was an accurate predictor and marker of radiation pneumonitis in rabbits led to further testing in the context of a clinical trial for patients with lung cancer. In that trial, surfactant apoprotein was measured in serum from blood samples collected before treatment, weekly during treatment, and at 1 week after treatment, and the findings were compared with the incidence and severity of radiation pneumonitis and fibrosis assayed by CT scans every 3 months after treatment and by chest radiography at 1 year after treatment.
Endothelial Cells The suggestion that vascular damage was the underlying mechanism of radiation pneumonitis and fibrosis was based primarily on the hypothesis that the lung cell most likely to divide soon after irradiation was the capillary endothelial cell. The parenchymal cells, particularly the type II cells, were thought to divide more slowly than endothelial cells. This hypothesis was substantiated by pathologic findings in humans and experimental models that pneumonitis after irradiation was characterized by edema in the air spaces and interstitium and an inflammatory cell infiltrate. Among the many investigators attempting to elucidate the role of the vasculature in radiation-induced lung damage, Ward et al.
86,
87,
88,
89 undertook comprehensive studies to correlate changes in four parameters of endothelial function (angiotensin-converting enzyme activity, plasminogen activator activity, and prostacyclin and thromboxane production) with histopathologic and ultrastructural changes in rat lungs irradiated with the same doses and with changes measured by arterial perfusion, a vascular functional assay. Those studies showed dose-dependent changes in all endothelial function parameters and good agreement between these functional changes and pathologic changes in the lungs of the rats. These findings suggest that changes in vasculature do occur after lung irradiation, but they do not necessarily implicate the endothelial cell as the target cell.
BIOLOGICAL BASIS OF RADIATION-INDUCED LUNG DAMAGE
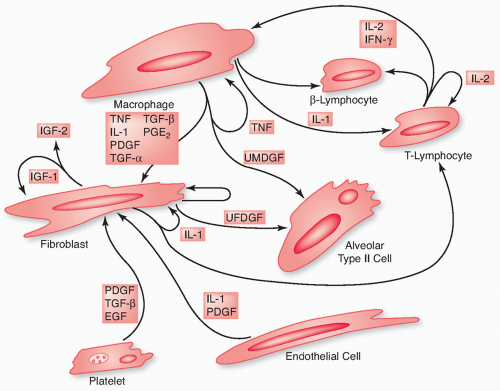
Changes in both the pulmonary parenchyma and pulmonary vasculature contribute to radiation pneumonitis and fibrosis (
Table 44.2), and these sequelae of lung irradiation arise from dynamic interactions among different cell types, the major players being type II pneumocytes and endothelial cells, as well as fibroblasts, macrophages, and lymphocytes (
Fig. 44.3).
90,
91 However, the concept that one or more target cells are solely responsible for radiation-induced pneumonitis and pulmonary fibrosis has been replaced by the paradigm that it is not only cells but also the messages that those cells send that determine the final outcome.
Molecular and Tissue Responses during Radiation-Induced Lung Damage Radiation is but one of many insults that prompt the release of trophic factors that subsequently act through various signaling pathways to
produce the pathologic end results of radiation pneumonitis and fibrosis.
92,
93,
94,
95,
96,
97 The messages are delivered via the diffusion of soluble mediators over short distances; such mediators may have been secreted by one population of cells and then act locally on either another population (paracrine stimulation) or the same population (autocrine stimulation), or they can interact with membrane-associated molecules that activate receptors on adjacent target cells (juxtacrine stimulation). These soluble mediators, known as cytokines or growth factors, are crucial in stimulating cells to overproduce the extracellular matrix components that characterize fibrosis.
23,
92,
93,
94,
98 After their initial contact with the cell surface, these molecules are postulated to activate intracellular signaling pathways, which in turn leads to activation of complex genetic programs characteristic of the fibrotic response.
Two concepts have been proposed to explain how cells read signals from cytokines.
93,
99,
100 In the first concept, cytokine signaling pathways are not insular or isolated paths or devices but rather are segments in a dense network that monitors itself through various crosstalk and feedback links and adjusts the activity of each constituent pathway. The nature of those adjustments, in turn, determines the nature and timing of the signals conveyed. In the second concept, the pathway provides the cell with information about the arrival of a certain cue but does not provide precise instructions. The cell, more than the pathway, determines the outcome of the signal.
100 In either event, cytokines are known to mediate the development of pneumonitis and fibrosis, both of which represent tissue responses to the damage induced by ionizing radiation. Several such cytokines, including transforming growth factor β (TGF-β), tumor necrosis factors (TNF-α and TNF-β), the interleukins, KL-6, and the intracellular adhesion molecule (ICAM)-1, are discussed further in the following paragraphs.
Transforming Growth Factor β The TGF-β cytokine family regulates the proliferation and differentiation of cells, embryonic development, wound healing, tumor progression, and angiogenesis
101,
102 and can trigger a bewildering diversity of responses depending on the genetic makeup and environment of the target cells.
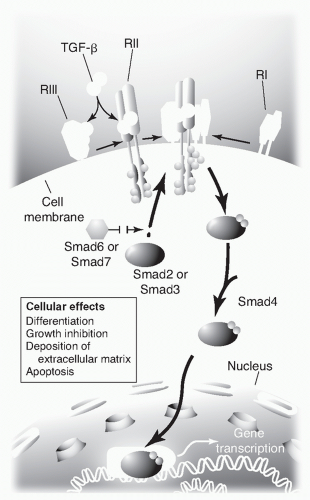
100 TGF-β regulates cellular process by binding to three high-affinity cell-surface receptors known as types I, II, and III (TGF-β: TGF-β1, TGF-β2, and TGF-β3). Each isoform is encoded by a distinct gene and is expressed in both a tissue-specific and a developmentally regulated fashion. The type I and II receptors contain serine-threonine protein kinases in their intracellular domains that initiate intracellular signaling by phosphorylating several transcription factors know as Smads (derived from the Sma and MAD gene homologues in
Caenorhabditis elegans and
Drosophila melanogaster). Of the 10 Smads, Smads 2 and 3 are phosphorylated by activated type I TGF-β receptors, whereas Smads 6 and 7 block the phosphorylation of Smads 2 and 3.
102 Deregulation of TGF-β signaling pathways has been implicated in the development of several major diseases, including cancer; atherosclerosis; fibrotic disease of the kidney, liver, and lung; and autoimmune diseases.
102 Mutations in the genes for TGF-β, its receptor, or intracellular signaling molecules associated with TGF-β are also important in the pathogenesis of disease, particularly cancer and hereditary hemorrhagic telangiectasia.
101
TGF-β is an important mediator of tissue damage in various abnormal conditions associated with excess collagen production. TGF-β1 in particular is often associated with lung fibrosis in response to several types of toxic insult, including radiation.
23,
98,
103 The TGF-β signal is transduced to the nucleus through the activation of its receptors, which in turn activate one or more Smad transcription factors, which then act on one or more of many potential target genes (
Fig. 44.4).
102,
104 In vivo, TGF-β stimulates the recruitment of lymphocytes and fibroblasts to sites of damage, promotes fibroblast proliferation, and stimulates the production of collagen and fibronectin— the effect being a net increase in extracellular matrix material, which eventually replaces the normal architecture of the tissue. TGF-β also increases the production of type I plasminogen activator inhibitor while simultaneously decreasing the production of plasminogen activators, resulting in not only an increase in the production of connective tissue but also a decrease in its breakdown, leading ultimately to maturation of this excess connective tissue and the formation of scars.
51,
102Rubin et al.
91 were the first to report that alveolar macrophages obtained from bronchial lavage specimens from irradiated rabbits demonstrated increased production and release of TGF-β relative to macrophages from normal lungs. Those investigators suggested that the fibroblast proliferation and extracellular matrix production seen after irradiation resulted from the release of cytokines such as TGF-β from parenchymal cells. Johnston et al.
105 reported an increase in the expression of TGF-β1 messenger RNA (mRNA) in radiation-fibrosis-prone but not in radiation-fibrosis-resistant strains of mice. That increase in TGF-β mRNA correlated with an increase in extracellular matrix protein expression in the irradiated lung tissue. Finkelstein et al.
92 studied whether changes in early expression of collagen precursors or cytokines known to be involved in the fibrotic process were expressed during the latent period, before damage was manifested histologically. These investigators used two single whole-thorax radiation doses to C57B1/6J mice, a strain known to be sensitive to the fibrogenic effects of lung radiation. The lower dose (5 Gy) is well below the threshold known to induce clinical symptoms and pathologic changes of radiation pneumonitis and fibrosis, and the higher dose (12.5 Gy) is closer to the reported LD
50 for radiation-induced lung damage in this strain, although this dose itself does not cause death from lung damage. The results showed that mRNAs for all three isoforms of TGF-β (i.e., TGF-β1, -β2, and -β3) were increased in the lungs of C57B1/6J mice at 14 days, but a consistent dose-response relationship was not found. Thus, radiation doses that cause neither clinical signs nor pathologic changes indicative of radiation lung damage can lead to changes in the expression of this fibrogenic cytokine before the onset of overt lung damage and may ultimately lead to the development of chronic fibrosis.
The importance of TGF-β1 in the development of radiation-induced lung damage has been underscored in human studies as well as in animal experiments.
106,
107,
108,
109 The feasibility of using TGF-β1 levels to identify patients who can safely tolerate radiation dose escalation was tested in 38 patients with non-small cell lung cancer (NSCLC).
106,
110 At the end of the initial radiation treatment, patients with TGF-β1 levels lower than pretreatment levels and patients with TGF-β1 levels of less than 7.5 ng/mL were selected for subsequent radiation dose escalation (86.4 vs. 73.6 Gy) to the primary tumor and enlarged lymph nodes. The predominant severe (grade ≥3) late toxicity in that study was not pulmonary but rather was esophageal, suggesting that monitoring plasma TGF-β1 levels might be useful for identifying patients resistant to both radiation-induced lung damage and esophageal toxicity.
106,
107,
110 In a later study, TGF-β1 levels were found to be elevated (>20 ng/mL) before treatment in 51% of 68 patients with NSCLC.
111 Plasma TGF-β1 levels after radiation therapy were associated with both pretreatment levels (
p=0.001) and with mean radiation dose to lung, and the mean ratio of plasma levels at weeks 4 to 6 of treatment to pretreatment levels predicted radiation pneumonitis, according to Southwest Oncology Group criteria (
p = 0.01) but not that defined by other scoring systems. However, this study was criticized for its high TGF-β1 baseline values, which were thought to be artifacts from the methods of blood collection and processing.
111Inflammatory Cytokines Just as depletion of one type of target cell is unlikely to trigger the complex pathologic process of radiation pneumonitis and fibrosis, it is equally unlikely that the overexpression of a single cytokine initiates and promulgates the fibrogenic process in the lung. Much more likely is that the damage, repair, and restructuring of the lung after irradiation (as well as after other toxic insults) are the results of a cytokine network orchestrated by a few key cytokines, such as TNF-α/β and TGF-β. Several classes of cytokines can contribute to the risk of radiation pneumonitis, among which the interleukins, TNF-α, and TGF-β1 have been recognized to play major roles. At the core of the inflammatory response after radiation exposure is the production of such proinflammatory cytokines as TNF-α and interleukin-1 and interleukin-6 (IL-1 and IL-6) and macrophages, lymphocytes, and other lung cell types.
112,
113,
114 Thus, it may be the balance of a few positive profibrogenic cytokines and negative antifibrogenic cytokines generated from the interaction of several cytokines constituting these networks that may finally determine the outcome of lung injury and inflammation. Unfortunately, dissecting the specific role of any of these cytokines in lung damage and repair is not easy; however, one approach to determining the role of various cytokines in lung fibrosis is to take advantage of known differences in susceptibility to radiation-induced fibrosis among inbred mouse strains.
Johnston et al.
115 used the fibrosis-prone C57B1/6J mouse strain and the fibrosis-resistant C3H/HeJ mouse strain to show that the elevation of mRNA levels of several chemokines implicated in the recruitment of inflammatory cells to sites of pulmonary damage was not different between the two strains when assessed at 8 weeks after irradiation. However, by 26 weeks after irradiation, messages encoding transcripts produced predominantly by macrophages and lymphocytes were elevated only in the fibrosis-prone mice. These findings indicate that lymphocytic recruitment and activation are key components of radiation-induced fibrosis, but their role in the pathogenesis of this process remains unclear.
Clinical studies have shown that elevation of IL-1α, IL-6, and IL-10 are associated with the risk of radiation-induced lung damage.
116,
117,
118 In fact, IL-1α and IL-6 have been used as markers for the diagnosis or prediction of radiation
pneumonitis.
118 TNF-α
119 vascular endothelial growth factor, and fibroblast growth factor (FGF) have also been shown to be associated with radiation lung injury.
120,
121,
122 Arpin et al.
116 found that IL-6 levels were significantly higher (
p= 0.047) during 3D conformal radiation therapy in patients who later were diagnosed with radiation pneumonitis and that covariations of IL-6 and IL-10 levels during the first 2 weeks of that therapy were independently predictive of radiation pneumonitis (
p =0.011). Chen et al.
118 also confirmed the importance of IL-6 measurements for the prediction of radiation pneumonitis. These results suggest that levels of one or more cytokines could be useful as biomarkers to monitor tissue response during the early course of radiation therapy with the goal of identifying patients who may benefit from adaptive redesign of that therapy.
Patient smoking status has also been linked with the risk of radiation pneumonitis. An in vitro study showed that cigarette smoke extract augmented the release of IL-8 from bronchial epithelial cells in a concentration-and time-dependent manner.
123 That study also showed that IL-8 in bronchoalveolar lavage samples from smokers was higher than that in samples from nonsmokers.
123 Other work has shown that exposure to tobacco smoke suppressed the expression of proinflammatory cytokine mediators.
124 In 2005, Hart et al.
125 reported that pretreatment IL-8 levels were four times higher in patients who did not develop symptomatic radiation-induced lung damage than in patients who did, suggesting that the IL-8 level might be useful for predicting who may develop radiation pneumonitis before the treatment is begun.
KL-6 The antigen KL-6 is a mucinlike high-molecular weight glycoprotein that is expressed on type II pneumocytes and bronchiolar epithelial cells and has been used as a serum marker of interstitial pneumonitis. The group that discovered KL-6 noted that 60% of patients with various kinds of interstitial pneumonitis had abnormally high KL-6 levels and that serum KL-6 levels correlated with the degree of clinical disease activity, as measured by gallium-67 citrate scintigraphy and the clinical course.
126 KL-6 also seems to be a useful marker for detecting severe pneumonitis and estimating its prognosis.
126,
127,
128 In another study of patients who developed severe radiation pneumonitis, serum KL-6 levels tended to increase before or after the clinical diagnosis of extensive radiation pneumonitis outside the radiation fields, but no change was noted before or after localized radiation pneumonitis inside the radiation fields. Moreover, patients whose serum KL-6 levels rose more than 1.5 times higher than their pretreatment serum KL-6 level were at greater risk of developing severe radiation pneumonitis that was unresponsive to steroid hormones and resulted in death.
129 In a study of women undergoing adjuvant radiation therapy for breast cancer, posttreatment serum levels were significantly different between patients with radiation pneumonitis and those without radiation pneumonitis.
130 Other investigators studying patients with primary and metastatic lung tumors who had been given single-fraction stereotactic radiotherapy found that the ratio of serum KL-6 levels at 2 months after treatment and levels at baseline correlated with the occurrence of radiation pneumonitis.
131 This 1- to 2-month gap between the time at which predictive measurements are the most accurate and the diagnosis of radiation pneumonitis, however, limits the predictive value of KL-6 for this purpose.
Intercellular Adhesion Molecule-1 ICAM-1 is an intercellular adhesion molecule of the immunoglobulin supergene family involved in adherence of leukocytes to the endothelium and in leukocytic accumulation in pulmonary injury. ICAM-1 is upregulated in association with inflammation in response to numerous types of inducing factors. In normal lung tissue, the expression of ICAM-1 on alveolar type I epithelial cells is stronger than on alveolar macrophages and on endothelial cells.
132 Selective ICAM-1 expression was detected on the surface of type I alveolar epithelial cells and, to a lesser extent, on the pulmonary capillary endothelium and on alveolar macrophages. Bleomycin-induced lung fibrosis demonstrated altered ICAM-1 distribution at the alveolar epithelial surface, and soluble ICAM-1 was detected by Western blot analysis.
132 The expression of ICAM-1 on pulmonary endothelial cells after stimulus and subsequent binding of neutrophils is a first step leading to lung injury. A similar process may dictate the binding of tumor cells to the pulmonary endothelium during metastasis. TNF-α upregulates ICAM-1 expression in a dose-and treatment interval-dependent fashion.
80
Expression of ICAM-1 can also be upregulated by gamma irradiation through the action of catalase. Furthermore, catalase, c-Jun N-terminal kinases (JNKs), and activator protein 1 (AP-1) activation induce ICAM-1 upregulation through a sequential process.
133 ICAM-1 and lymphocyte function-associated antigen-1 (LFA-1) expression on alveolar macrophages was significantly increased starting 1 week after irradiation, whereas their expression on lung tissue was not elevated up to 8 weeks after irradiation, suggesting that adhesion molecules play a role in the development of radiation-induced lung injury.
134 Using immunohistochemical assay for upregulation of adhesion molecules associated with recruitment, transendothelial migration, and proliferation of bronchoalveolar macrophages, researchers found significant upregulation of vascular cell adhesion molecule-1 (VCAM-1) and ICAM-1 at 100 days after 20 Gy to total lung irradiation with further increases up to the time of death. Increases were first detected in endothelin-positive endothelial cells. These data suggest an association between late irradiation-induced pulmonary fibrosis and the upregulation of adhesion molecules and disclose potential targets for intervention in the pulmonary vascular endothelium.
135Clinically, expression of human leukocyte-associated antigen (HLADR) and ICAM-1 on T cells in bronchoalveolar lavage fluid increased in patients who developed radiation pneumonitis after radiation for lung cancer. A significant association was seen between the incidence of ICAM-1 expression on T cells and the number of days from the initiation of radiotherapy to the onset of radiation pneumonitis.
136 In another study, 30 patients were irradiated with a total dose of approximately 60 Gy. Blood samples were taken before, midway, and after radiotherapy.
Bronchoalveolar lavage was also performed before and after radiation therapy in seven cases. A total of 12 out of 30 (40%) cases developed radiation pneumonitis. Serum levels of soluble ICAM-1 after radiation therapy were significantly elevated in patients who developed pneumonitis compared with those who did not. In some of the cases, sICAM-1 levels began to increase at an early phase of irradiation. These findings suggest that ICAM-1 may have an important role in the development of radiation pneumonitis and that sICAM-1 may be a useful marker for the early detection of radiation pneumonitis.
137Thus, despite abundant evidence that expression of a multitude of cytokines is elevated in radiation-induced pneumonitis and lung fibrosis, their actual in vivo role and the mechanism of their upregulation remain to be determined. Regardless of our lack of understanding of how these factors mediate damage and repair in the lung, it is clear that the latent phase preceding the overt expression of radiation damage in the lung is not a quiescent time. Molecular changes are occurring that may later ultimately result in the clinical and histopathologic picture of pneumonitis and fibrosis. The events that lead to the overt expression of radiation-induced lung damage involve several complex cellular and molecular processes, which explain why both the inflammatory responses and the triggering mechanism that induces the fibrotic response in the lung remain poorly understood.
Genetic Regulation
Animal Studies Animal studies, particularly those showing strain-dependent outcomes, indicate that genetics are implicated in vulnerability to pneumonitis and fibrosis. Twenty years ago, Steel et al.
138 reported an anomalous finding after irradiation of the whole thorax of C57B1 mice that the survival time of this inbred strain of mice was significantly longer than the standard 80 to 180 days reported in other strains with radiation pneumonitis. Subsequent studies by Down et al.
139,
140 in several inbred mouse strains showed that the incidence of radiation pneumonitis and fibrosis was strain dependent. Although strain-dependent differences could result from several factors, genetics offers a plausible explanation. In an in-depth survey of the incidence of radiation pneumonitis and fibrosis in nine inbred mouse strains, Sharplin and Franko
43 showed that the strains could be categorized as fibrosing, nonfibrosing, or intermediate, based on quantitative histologic findings. Further studies by these authors showed that crossbreeding strains with different proneness to fibrosis produced hybrids with the same sensitivity of the parent least prone to fibrosis: the hybrid of a fibrosing and a nonfibrosing strain was uniformly nonfibrosing, whereas a cross of an intermediate with a fibrosing strain produced an intermediate hybrid,
141 suggesting that sensitivity to radiation-induced fibrosis is an autosomal recessive trait. Supporting this hypothesis is a report from Haston et al.
51 that one locus on chromosome 17 segregates with the fibrosing phenotype and that this locus, which is within the region containing the major histocompatibility locus (MHC), was linked to both bleomycin-induced lung fibrosis
142 and susceptibility to ozone-induced lung damage in mice.
143 These researchers also showed that a quantitative trait locus on chromosomes 17 influenced susceptibility to radiation-induced pulmonary fibrosis. Chromosome 6 (logarithm of the odds [LOD]= 4.6), which has an additional region containing a quantitative trait locus, showed linkage in female mice only. The evidence for linkage to chromosome 18 weakened when it was analyzed jointly with other markers. These loci—on chromosomes 1, 6, 17, and 18—were estimated to account for 70% of the genetic contribution to this trait, with chromosome 17 accounting for 28% and chromosome 1 for 24%. Furthermore, the quantitative trait locus on chromosome 17 for radiation-induced lung fibrosis is within the same region as a quantitative trait locus identified for lung damage after other insults (e.g., from bleomycin, ozone, and particle exposure) as well as from asthma, suggesting that this region of chromosome 17 may harbor a “universal” lung injury gene.
144 These findings suggest that although other loci have been shown to be insult dependent, at least one genetic factor regulating pulmonary fibrosis may be universal and independent of the etiology of that fibrosis.
Also supporting a genetic sensitivity theory is that other pathologic processes in the lung are known to be under genetic regulation. Pulmonary inflammation is well-known to be controlled by genes in the H-2 complex.
145,
146,
147 Mice with an H-2
k locus are high responders, and those with an H-2
b locus are low responders.
145,
147,
148 In the studies of Sharplin and Franko
43 and Down et al.,
140 pneumonitis was found to be strain dependent and related to the H-2 locus; that is, an inflammatory cell infiltrate was not a feature of pneumonitis in the highly fibrogenic strain, and, conversely, an inflammatory cell infiltrate was the most prominent characteristic of the pneumonitis phase in the weakly fibrogenic strain. Although these observations refute the hypothesis that immune-inflammatory processes usually precede fibrosis, they clearly indicate that pneumonitis after irradiation of mouse lung is genetically regulated. However, the strain sensitivity to radiation pneumonitis is exactly opposite for that of radiation-induced fibrosis.
Two forms of lung fibrosis have been described.
31 One form of fibrosis is a collagen deposition in the interstitium and air spaces, resulting in contracture and collapse of the alveoli, an obliteration of normal lung architecture, and an attendant loss of pulmonary function resulting in the death of the mouse. Another form of fibrosis is characterized by collagen deposited only in the interstitium, a process that maintains alveolar structure, and was largely ignored because it was suggested that this lesion alone would not produce sufficient functional impairment to cause death. The genetic regulation that controls these two forms of fibrotic processes may not be identical to that controlling interstitial fibrosis, although there may be overlap between the two. Furthermore, interstitial fibrosis in the lung
18 and colon
53 can result in death in animal studies.
139Evidence suggests that the fibrosis evident in mouse lung after exposure to radiation can arise through two independent mechanisms: one is controlled by two autosomal recessive
determinants that act additively, and the other is regulated independently by two additional genes, one of which is X-linked.
45 Similar strain variations in pulmonary fibrosis after other kinds of insults have been reported. Particularly interesting are the results obtained after treatment with bleomycin, another DNA-damaging agent often used to induce lung fibrosis in model systems. The most striking observation after bleomycin treatment is that
149,
150,
151,
152 the findings were remarkably similar to those observed after radiation. These findings, like those reported by Haston et al.,
144 suggest that genetic factors related to susceptibility to lung fibrosis may operate independently of the etiologic agent.
Clinical Evidence Intrinsic sensitivity to radiation is known to differ among individuals. Estimates indicate that, given the same standardized radiation dose, technical and clinical factors account for about one third of the interpatient variation in normal-tissue reactions, and genetic differences between patients account for the greater proportion of differences in sensitivity.
153 Certainly the severity of treatment-related complications, including the severity of radiation-induced pneumonitis and fibrosis after definitive radiation therapy for lung cancer, have been evident clinically for some time. Geara et al.
154 demonstrated considerable patient-to-patient heterogeneity in a group of 56 patients with limited small cell lung cancer treated with chemoradiation therapy, suggesting that the risk of lung fibrosis is strongly affected by inherent factors that vary among individuals.
Evidence is increasing that indicates that genetic variations in a few selected cellular pathways, such as those involved in DNA repair, cell cycle, and inflammation, may affect radiation sensitivity, treatment response, and toxicity.
155,
156,
157,
158,
159,
160,
161,
162 For example, patients with the genetic syndromes of ataxia telangiectasia, Nijmegen breakage syndrome (NBS1), or Bloom syndrome (BLM) demonstrate hypersensitivity to radiation,
163,
164,
165 and genes that are defective in these syndromes (ataxia telangiectasia mutated [ATM], NBS1, or BLM, respectively) are known to play critical roles in DNA repair, particularly double-strand break repair. In vitro studies in cell lines that are hypersensitive to radiation have demonstrated that the repair genes for double-strand breaks, x-ray repair cross-complementing 1 gene (XRCC1) to x-ray repair cross-complementing 7 genes (XRCC7), are responsible for hypersensitivity. Patients with immune deficiencies arising from defects in double-strand break repair genes, such as defects in the DNA-dependent protein kinase catalytic subunit (DNA-PKcs) (XRCC7), Ku70 (XRCC6), and Ku86 (XRCC5) genes, exhibit high radiation sensitivity.
166,
167,
168Family studies also suggest a genetic predisposition to radiosensitivity. Roberts et al.
169 studied the heritability of radiation sensitivity in peripheral blood lymphocytes in families of patients with breast cancer. They found that 62% of the first-degree relatives of radiation-sensitive patients were also sensitive compared with 7% of the first-degree relatives of patients with normal sensitivity. Segregation analysis of 95 family members showed clear evidence of high heritability of radiation sensitivity.