Respiratory Pump Failure |
Pump Failure: The Pathogenesis of Hypercapnic Respiratory Failure in Patients with Lung and Chest Wall Disease
The ventilatory pump accomplishes bulk transfer of air to and from the alveoli. Accordingly, diseases that perturb the mechanical properties of any component of the ventilatory pump (i.e., the bony rib cage, the extra- and intrathoracic conducting airways, and the respiratory muscles) may interfere with CO2 elimination and O2 uptake. If disturbances in the function of the ventilatory pump are sufficiently severe, alveolar hypoventilation and respiratory acidosis may ensue. Hypercapnic respiratory failure is defined as a steady-state PaCO2 while awake at more than 45 mm Hg, the upper limit of normal. This definition is somewhat arbitrary but has proved clinically useful.
Conceptually, diseases that cause hypercapnic respiratory failure do so by deranging respiratory mechanics and lung dead space volume (e.g., chronic obstructive pulmonary disease [COPD], asthma, or kyphoscoliosis) or by impairing the contractile properties of the respiratory muscles (e.g., neuromuscular disease). Diseases that impair respiratory mechanics increase the elastic or resistive load against which the respiratory muscles must contract. On the other hand, neuromuscular diseases impair the strength or endurance properties of the respiratory muscles and impair their ability to generate swings in intrathoracic pressure sufficient to maintain ventilation.
The rhythmic act of breathing results from the activity of a central respiratory pattern generator (CRPG) comprises interacting networks of excitatory and inhibitory neurons in the pons and medulla oblongata.1,2 In turn, projections from the CRPG to bulbospinal motor neurons activate the respiratory skeletal muscles in the chest wall, abdomen, and upper airway and, hence, shape the neuromuscular drive to breathe. A variety of compensatory neural mechanisms located in the periphery that sense alterations in blood-gas tensions or ventilatory performance which project to the CRPG elicit increases in the neuromuscular drive to breathe and, in turn, help preserve alveolar ventilation.3–7 In fact, in most patients, rather marked abnormalities in ventilatory pump performance are required before hypercapnic respiratory failure ensues. Conceptually, the susceptibility to develop CO2 retention in the setting of lung, chest wall, or respiratory muscle dysfunction, therefore, depends on the balance between the severity of the derangement in ventilatory pump function and the intensity of the respiratory neuromuscular drive to breathe.5
This chapter deals with the pathogenic mechanisms at work in the development of CO2 retention in lung and chest wall diseases. The compensatory/adaptive mechanisms that help preserve ventilation (e.g., respiratory chemosensitivity, motor responses to alterations in the mechanics of breathing, and intrinsic changes in respiratory muscle strength and endurance) and the decompensating/maladaptive responses that predispose to CO2 retention (e.g., respiratory muscle wasting and fatigue and a rapid, shallow pattern of breathing) will be discussed.
COMPENSATORY/ADAPTIVE MECHANISMS
The roles of compensatory or adaptive mechanisms in hypercapnic respiratory failure are considered with respect to respiratory chemosensitivity, blunted chemosensitivity, and alterations in respiratory structure.
 RESPIRATORY CHEMOSENSITIVITY
RESPIRATORY CHEMOSENSITIVITY
Below is a discussion of normal respiratory chemosensitivity and measures of respiratory motor output.
Overview—Regulation of Ventilation
Hypoxia and hypercapnia stimulate chemoreceptors in the arterial circulation (peripheral chemoreceptors) and ventrolateral medulla (central chemoreceptors) that reflexively increase motor activity to the respiratory skeletal muscles of the chest wall and upper airway.8 Contraction of the muscles of the chest wall (e.g., diaphragm, intercostals, abdominals, and neck muscles) deforms the ventilatory pump and moves air.5 Contraction of the muscles of the upper airway (genioglossus, alae nasi, posterior arytenoids, pharyngeal dilators, sternohyoid, etc.) increases the caliber of the upper airway and diminishes its susceptibility to collapse during inspiration.6
Chemoreceptor-induced increases in inspiratory and expiratory muscle activity are proportional to the severity of abnormalities in blood-gas tensions and represent a feedback control loop that restores blood-gas tensions toward normal by enhancing alveolar ventilation. The magnitude of the swings in intrathoracic pressure and resistance and compliance of the upper airway are determined by these changes in respiratory motor activity. The maintenance of blood-gas tensions within a relatively narrow, normal range from neonatal life to senescence attests to the power of this homeostatic mechanism.
Hypoxic and hypercapnic chemical drives to breathe exert the following stereotypic effects on the activity of chest wall and upper airway muscles.6,8,9 Peak respiratory muscle electrical activity and its rate of rise are increased. For the inspiratory muscles, these changes in muscle electrical activity increase the rate of change and peak inspiratory intrathoracic pressure, inspiratory airflow, and tidal volume. For the expiratory muscles, increased electrical activity enhances the rate of expiratory airflow. For the upper airway muscles, the resistance to inspiratory airflow decreases.
Chemosensitivity-induced increases in respiratory activity also affect the timing of respiratory motor activity as reflected in the duration of inspiration (TI) and expiration (TE).10,11 Hypoxia and hypercapnia lead to decreased TI and TE, allowing the frequency of breathing to increase. Reductions in TE are generally out of proportion to TI, thereby increasing the fraction of the respiratory cycle spent in inspiration. This partitioning of the respiratory cycle is reflected in the TI/TT ratio, where TT is the total breath cycle duration (i.e., the sum of TI and TE).
Hypoxia and hypercapnia differ in their effects on the activity of the inspiratory muscles after the cessation of inspiratory airflow, the so-called postinspiratory inspiratory activity (PIIA).1 Hypoxia increases PIIA in both chest wall inspiratory muscles and muscles that constrict the laryngeal aperture. Accordingly, hypoxia has a braking effect on the rate of expiratory airflow. As TE decreases with increasing hypoxic drive, end-expiratory lung volume increases. PIIA-induced increases in lung volume increase the caliber of the intrathoracic airways and the O2 content of the lung. Hypoxia-induced PIIA affects the load on the respiratory muscles in complex fashion; that is, PIIA reduces inspiratory resistive work of breathing but increases the inspiratory elastic and expiratory resistive work of breathing. It has been suggested, however, that the net effect of hypoxia-induced PIIA is a reduction in overall energy expenditure during breathing. In contrast, hypercapnia diminishes the duration of PIIA.
Indices of Respiratory Motor Output
Ventilation is a well-accepted index of respiratory motor output.8,10–12 Traditionally, ventilation was viewed as the product of tidal volume (VT) and respiratory rate (which is equal to 60/TT). More recently, ventilation has been viewed as the product of separate “drive” and “timing” components.10 The average rate of inspiratory airflow, VT/TI – which reflects the rate of rise of inspiratory muscle activity and intrathoracic pressure – is increased when blood-gas tensions are deranged. Accordingly, VT/TI has been taken as a reflection of the activity of mechanisms that regulate the drive to breathe. Of note, VT/TI may also be increased by excitatory inputs arising from respiratory mechanoreceptor afferents (e.g., vagal irritant receptors) and higher central nervous system (CNS) structures engaged in thermoregulation and emotion (i.e., hypothalamic and limbic areas). Conversely, the TI/TT ratio has been taken as a reflection of the activity of mechanisms that regulate respiratory timing. The TI/TT ratio is strongly affected by afferent input from mechanoreceptors in the lungs, airways, and respiratory muscles, as well as increasing chemical drive. For example, TI/TT increases in anesthetized animals when vagal stretch receptors are stimulated by increases in lung volume and is decreased by bronchoconstriction-induced activation of vagal irritant receptors.
In subjects with normal lung function, VT/TI and ventilation are accurate reflections of inspiratory muscle electrical activity and the rate of rise of intrathoracic pressure. On the other hand, diseases that adversely affect the mechanical properties of the ventilatory pump (e.g., obstructive lung disease, kyphoscoliosis) interfere with the translation of changes in intrathoracic pressure into ventilation and airflow. Conversely, conditions that impair respiratory muscle contractility (e.g., neuromuscular diseases, respiratory muscle fatigue) interfere with the translation of inspiratory muscle electrical activity into intrathoracic pressure changes. Accordingly, VT/TI reflects the intensity of motor outflow to the inspiratory muscles produced by increasing chemical drive only when the mechanical properties of the ventilatory pump and inspiratory muscle strength are normal. When the ventilatory pump function is abnormal, respiratory motor outflow is best assessed from respiratory muscle electrical activity (i.e., diaphragm electromyography [EMG] activity), a complicated measurement largely confined to the research laboratory.
A simpler, clinically useful measurement that reflects the neuromuscular drive to breathe and the driving pressure to inspiratory airflow is the airway occlusion pressure.10,13–15 The occlusion pressure is the pressure generated at the airway opening 100 ms after the onset of an occluded inspiratory effort (i.e., P100 or P0.1) initiated at end-expiratory lung volume. Since the airway is occluded, the inspiratory muscles contract quasi-isometrically, a condition in which muscle force correlates closely with muscle electrical activity. Measurements are made early in inspiration (100 ms) to prevent behavioral responses elicited in response to airway occlusion from altering the shape/trajectory of the pressure waveform. The lack of flow or volume change during the measurement means that the occlusion pressure is unaffected by abnormalities in the flow-resistive or compliance properties of the ventilatory pump. The occlusion pressure, therefore, has been used to assess the drive to breathe in patients with lung diseases (e.g., COPD and asthma) and chest wall diseases (e.g., kyphoscoliosis) during resting and chemically stimulated breathing. On the other hand, the occlusion pressure depends on the ability of the inspiratory muscles to convert neural activity into force and pressure. Accordingly, like ventilation, occlusion pressure may not reflect respiratory motor neuron activity when the inspiratory muscles are weak (e.g., neuromuscular disease) or fatigued.
Hypoxic Response Under isocapnic conditions, ventilation (or occlusion pressure) increases in curvilinear fashion as PO2 falls.8 However, hypoxic responses depend importantly on the prevailing level of PaCO2 (i.e., the O2–CO2 interaction). When PaCO2 is in the hypocapnic range, arterial PO2 must fall considerably (to approximately 55–60 mm Hg or less) before respiratory activity increases. Hypercapnia profoundly increases the response to hypoxia by shifting the threshold of the response toward higher levels of PO2 and augmenting the change in ventilation elicited for a given reduction in PO2.
Although the physiological stimulus for the hypoxic response is the PaO2 of the blood perfusing the peripheral chemoreceptors, for convenience the oxyhemoglobin saturation assessed with a pulse oximeter has been taken as a reflection of the stimulus. Use of the oxyhemoglobin saturation linearizes the relationship between the hypoxic stimulus, ventilation, and occlusion pressure. The intensity of the hypoxic response has been assessed from the slope of the change in ventilation (or occlusion pressure) relative to the change in O2 saturation (i.e., ΔVE/Δ% O2 sat) and from the intercept of the relationship (e.g., ventilation at O2 saturation of 85%).
Hypercapnic Response In contrast to the response to hypoxia, the ventilatory and occlusion pressure responses to hypercapnia under iso-oxic conditions are linear over a relatively wide range of PaCO2 above and below the resting level of 40 mm Hg.8 The intensity of the ventilatory and occlusion pressure response to CO2 has been assessed from the slope of the relationship of VE to PaCO2 (i.e., ΔVE/ΔPaCO2) and from the intercept of the relationship (i.e., VE at PaCO2 50 mm Hg).
The ventilatory response to hypercapnia is strongly affected by the prevailing level of PaO2 and is heightened as PaO2 decreases. In fact, hypoxemic and hypercapnic stimuli interact multiplicatively to enhance inspiratory and expiratory motor activities. Worsening hypoxemia enhances the ventilatory response to hypercapnia in accordance with the O2–CO2 interaction. The strength of a subject’s chemosensitivity to O2 and CO2 and, in particular, to the O2–CO2 interaction is a powerful feedback mechanism opposing the tendency to retain CO2 in patients with ventilatory pump dysfunction.
Consequently, treatment of the hypercapnic, hypoxemic patient with supplemental O2 may decrease VT/TI and TI/TT and, hence, worsen hypercapnia in accordance with O2–CO2 interaction. Increases in PaO2 in hypoxic, hypercapnic subjects move the O2 response to the right (less stimulus) and decrease the slope and shift the intercept of the ventilatory response to hypercapnia to the right (Fig. 143-1). Shifts in the CO2 response with increases in the prevailing PaO2 mean that a higher CO2 stimulus is required to maintain ventilation at the baseline level. Accordingly, ventilation falls and PaCO2 rises. The magnitude of the rise in PaCO2 in patients with COPD in acute respiratory failure produced by supplemental O2 varies widely among subjects as determined by their chemosensitivity.
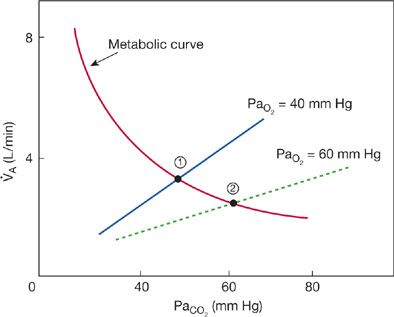
Figure 143-1 Theoretical effects of supplemental O2 on the ventilatory response to CO2 and steady-state arterial PaO2 in subjects with COPD in hypercapnic respiratory failure. Increasing PaO2 decreases alveolar ventilation and increases PaCO2 as dictated by effects of O2 on the CO2 ventilatory response. The two straight lines represent hypercapnic ventilatory response curves at PaO2 of 40 and 60 mm Hg. As may be seen, increasing PaO2 produces a downward, rightward shift of the ventilatory response. In contrast, the hyperbolic line intersecting the ventilatory response lines is the metabolic CO2–ventilation curve, which represents the effect of increasing alveolar ventilation (independent variable) on PaCO2 (the dependent variable) when CO2 production is normal (~200 mL/min). Steady-state alveolar ventilation and PaCO2 at rest are dictated by intersection of the ventilatory response curves with the metabolic curve (points 1 and 2). Note the increase in PaCO2 as the ventilatory response with PaO2 60 mm Hg intersects at a lower alveolar ventilation and higher PaCO2 (point 2) compared to the higher ventilatory response when PaO2 was 40 mm Hg (point 1).
Of note, hypercapnia induced by supplemental O2 in patients with COPD is multifactorial and reflects increases in lung dead space volume as well as reductions in alveolar ventilation. Hypoxemia causes bronchoconstriction via increases in parasympathetic outflow to airway smooth muscle. Accordingly, relief of hypoxemia causes bronchodilation and increased dead space volume.
 ROLE OF BLUNTED CHEMOSENSITIVITY IN DEVELOPMENT OF RESPIRATORY FAILURE
ROLE OF BLUNTED CHEMOSENSITIVITY IN DEVELOPMENT OF RESPIRATORY FAILURE
Chemosensitivities to hypoxemia and hypercapnia are heredofamilial and ethnic traits that vary widely interindividually (Fig. 143-2).16,17 In a given subject, responses to hypoxemia and hypercapnia correlate weakly, so that subjects with strong responses to hypercapnia also tend to have strong responses to hypoxia. Respiratory chemosensitivity to both hypoxemia and hypercapnia declines with age.18 The decline in chemosensitivity with aging may explain why elderly subjects with lung disease (e.g., COPD) or chest wall disease (e.g., kyphoscoliosis) develop hypercapnic respiratory failure more frequently than young adults. When chemosensitivity is low, subjects with diseases of the ventilatory pump are predisposed to develop hypercapnic respiratory failure.16,17,19
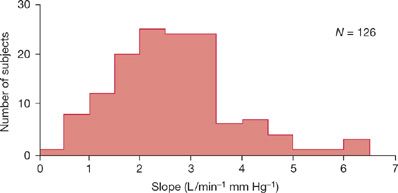
Figure 143-2 Variability of the slopes of the ventilatory responses to progressive hypercapnia (i.e., PE/PCO2) in a normal population. Shown is the frequency distribution histogram of the slopes in 126 normal South African medical students. Note the considerable interindividual variation in CO2 responsiveness. In some healthy subjects, the ventilatory response is blunted to less than 1 L/min/mm Hg PCO2. (Data from Irsigler GB. Carbon dioxide response lines in young adults: The limits of the normal response. Am Rev Respir Dis. 1976;114:529–536.)
In patients with advanced COPD, the severity of airway obstruction required to cause CO2 retention varies widely from subject to subject (Fig. 143-3).20,21 Subjects with the greatest respiratory effort responses to changes in PaCO2 – as measured by diaphragm EMG, respiratory work of breathing, or occlusion pressure – have arterial PaCO2 values closer to normal than subjects with blunted responses to CO2 but the same severity of lung dysfunction. Accordingly, when chemosensitivity is low, subjects with diseases of the ventilatory pump are predisposed to develop hypercapnic respiratory failure. However, since CO2 retention per se may blunt the response to acute hypercapnia, studies in patients in respiratory failure have not been able to determine whether blunted CO2 responses are a cause or consequence of respiratory failure.22
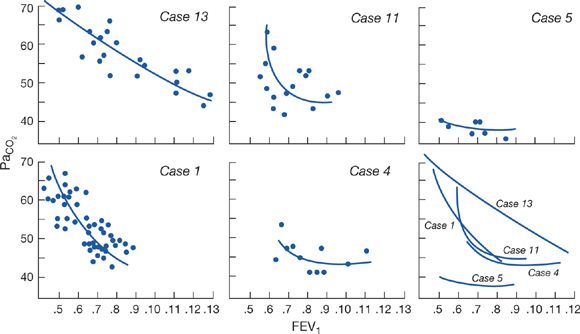
Figure 143-3 Results of repeated measurements of arterial PCO2 and FEV1 (liters) in five patients with advanced COPD. Free-hand curves of the data are shown plotted together in the lower right graph. Note that cases 1, 11, and 13 show marked increases in PCO2, with relatively small changes in FEV1, whereas cases 4 and 5 do not. (Data from Lane DJ, Howell JBL, Giblin B. Relation between airways obstruction and CO2 tension in obstructive airways disease. Br Med J. 1968;3:707–709.)
The tendency for chemosensitivity to be inherited has been used in a number of subsequent studies to assess the role of hypoxic and hypercapnic responses in the pathogenesis of CO2 retention in the setting of obstructive lung disease. Study of relatives with normal lung function and blood gases has been employed to circumvent the effects of CO2 retention on respiratory chemosensitivity in patients with COPD.
In general, normal relatives of hypercapnic patients with COPD have lower ventilatory responses to hypoxia and hypercapnia than relatives of eucapnic patients with COPD (Fig. 143-4).17 The slopes of the ventilatory responses to isocapnic hypoxemia and hyperoxic hypercapnia are 30% to 40% lower in the offspring of hypercapnic patients than in the offspring of eucapnic patients. Similarly, the slopes of the ventilatory and airway occlusion pressure responses to isocapnic hypoxia are approximately 40% lower in the offspring of hypercapnic patients than in the offspring of normocapnic patients. In one study, the PaO2 of COPD patients while in a stable state and the PaO2 and PaCO2 during COPD exacerbations correlated with the hypoxic ventilatory response of their sons. It appears that blunted chemosensitivities23 to hypoxia and hypercapnia are likely to be premorbid characteristics of hypercapnic patients with COPD, which contribute to the development of respiratory failure.
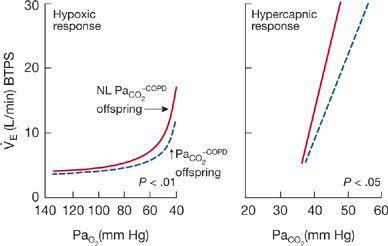
Figure 143-4 Mean isocapnic hypoxia and hyperoxic hypercapnic ventilatory response curves of 12 offspring of hypoventilating patients with COPD (solid line) and 10 offspring of eucapnic COPD patients (dashed line). Ventilatory responses to hypoxia and hypercapnia are significantly lower in the offspring of hypercapnic COPD than in the offspring of eucapnic COPD patients. (Data from Mountain R, Zwillich CW, Weil JV. Hypoventilation in obstructive lung disease: The role of familial factors. N Engl J Med. 1978;297:521–525.)
A number of reports describe patients with asthma and respiratory failure who had blunted ventilatory responses to hypoxia and hypercapnia and whose healthy immediate family members also showed blunted hypoxic and hypercapnic responses. Respiratory responses to hypoxia and hypercapnia in patients with asthma who have near-fatal attacks differ from those of asthmatics who did not have near-fatal attacks and age-matched, normal subjects.19 The slopes of the ventilatory and occlusion pressure responses to hypoxia in the patients with a history of near-fatal asthma are approximately 33% similar. Hypercapnic responses tend to be lower in the near-fatal asthmatic groups than in the other two groups, but the differences are smaller in magnitude. Of interest, recent studies indicate that hereditary loss of function mutations in the neuronal transcription factor, paired-like homeobox 2B (Phox2b), which are required for normal development of the retrotrapezoid nucleus, a chemosensitive region, are associated with impaired chemosensitivity to hypercapnia, resting hypercapnia and central hypoventilation in man.23,24
Responses to Heightened Respiratory Load
A complex array of mechanoreceptors and proprioceptors whose afferents project to respiratory neurons in the brain stem and higher CNS structures provides the respiratory controller with information about the mechanics of breathing and performance of the ventilatory pump.6 The sensory receptors providing this afferent feedback and the CNS structures that integrate this feedback into a coordinated respiratory response are not perfectly understood. However, mechanoreceptors in the intercostal muscles that sense muscle tension and length (Golgi tendon receptors and spindle organs, respectively) and pressure and flow sensors in the lower (vagal irritant receptors) and upper airway (larynx and mouth) clearly play a role in shaping the neuromuscular response to alterations in the mechanics of breathing.
Diseases of the airways (COPD and asthma) or chest wall (kyphoscoliosis) change the resistance and compliance properties of the ventilatory pump and, hence, stimulate mechanoreceptors in the ventilatory pump. In normal subjects and those with COPD, mechanoreceptor afferent inputs increase inspiratory neuromuscular output as reflected in airway occlusion pressure in response to bronchoconstriction or external resistances or elastance. Changes in ventilation during acute increases in airway resistance are inversely related to changes in occlusion pressure. Thus, the magnitude of the motor response to increases in respiratory load determines the ventilatory response.
External ventilatory loads that can be consciously detected and alter the intensity of the sensations associated with breathing elicit increases in respiratory effort as reflected by the diaphragm EMG and occlusion pressure.3,25 Increases in effort occur abruptly within the first loaded breath and in feed-forward fashion; that is, the experience of the previous breath elicits a response in anticipation that the load will still be present. These responses are eliminated by general anesthesia and dulled if not absent in stages III and IV and REM sleep. The afferent input to the CNS elicited by external ventilatory loads probably arises from spindle and tendon organs in the respiratory muscles that project to the sensorimotor cortex and medullary respiratory neurons. The motor response to external ventilatory loads is thought to be behavioral.
The magnitude of the respiratory motor response to external loads varies widely from subject to subject and may be a heredofamilial trait. Of considerable importance, some subjects with COPD demonstrate lesser occlusion pressure responses to acutely applied external resistive loads than age-matched normal subjects.22 It has been suggested that the blunted respiratory motor response to external loads may be a form of sensory adaptation to chronic increases in respiratory resistance. The fact that occlusion pressure responses of patients with COPD to external elastic loads and patients with asthma to external resistive loads are normal supports this concept. Of interest, the blunted motor response to external loads in some patients with COPD may reflect an increase in endogenous opiates within the CNS, since naloxone administration immediately enhances the response.
In subjects with COPD, bronchoconstriction increases airway occlusion pressure in proportion to increases in airway resistance and to a greater extent than external flow–resistive loads.26 Bronchoconstriction increases the activity of vagal “irritant” receptors in the airway, which exert an inspiratory augmenting effect on breathing. Irritant receptors may also be excited chemically by inflammatory mediators (e.g., histamine, prostaglandin F2α) and, in contrast to external loads, elicit simple monosynaptic reflexes not abolished by sleep or anesthesia.
Mechanoreceptor inputs modify the respiratory motor responses to chemical stimuli to breathing. Increases in the elastic or resistive load to inspiration augment inspiratory muscle electrical activity and the airway occlusion pressures to hypoxia and hypercapnia. Subjects with asthma show heightened occlusion pressure responses to hypoxia and hypercapnia for this reason. Increases in the inspiratory neuromuscular drive to breathe allow ventilation to be maintained in the face of abnormalities in respiratory mechanics. Respiratory motor activity (i.e., occlusion pressure) also tends to be increased when the respiratory muscles are weak. In all likelihood, this reflects the fact that the maintenance of force output by a weakened muscle requires an increase in activation by the CNS.
Increased ventilatory loads also alter the pattern of breathing in load-dependent fashion.3 Subjects breathing against resistive loads breathe slowly and deeply, with an increase in tidal volume and prolongation of TI and TE. In contrast, subjects breathing against elastic loads tend to breathe with smaller tidal volumes and a reduced TI and TE; that is, they demonstrate a rapid and shallow pattern of breathing. Slow, deep breathing during resistive loading and rapid, shallow breathing during elastic loading diminish the resistive and elastic work of breathing, respectively. Alterations in breathing pattern when the mechanics of breathing are deranged are believed to be attempts to minimize the work of breathing, muscle tension, or energy expended.
Integrated Motor Responses
Respiratory motor responses to heightened chemical or mechanoreceptor drives to breathe elicit highly coordinated patterns of muscle activity that optimize the mechanical output of the respiratory musculature contracting in concert.5,6,27,28 These responses may take the following forms1: Simple reflex-mediated recruitment of additional agonists, which exert similar mechanical effects on the chest wall2; sequential activation of inspiratory and expiratory muscles, which exert opposing effects on chest wall structures; and3 complex behavioral acts that use nonrespiratory muscles to effect changes in body posture and expiratory airflow, minimizing dyspnea.
For example, hypercapnia and hypoxia recruit the external intercostal and parasternal muscles during inspiration in a stereotypic rostral-to-caudal direction, and the internal intercostals and triangularis sterni during expiration in the opposite direction. Preferential activation of the inspiratory external intercostal and parasternal muscles in the rostral-most interspaces decreases the impedance of the rib cage to rostral movement and, hence, facilitates thoracic expansion. Conversely, recruitment of the expiratory internal intercostals and triangularis sterni in the most caudal interspaces decreases the impedance to caudal movement and facilitates thoracic deflation. In addition, recruitment of the parasternal intercostal muscles facilitates inspiratory pressure as tidal volume increases. The parasternal intercostal muscle fiber length, which is optimum for tension development, is shorter than that of the diaphragm and occurs at higher lung volume. Accordingly, the parasternal muscles become mechanically more effective than the diaphragm as lung volume increases above functional residual capacity (FRC).
Moreover, hypercapnia and hypoxia increase phasic and tonic inspiratory activities in the dilator muscles of the upper airway (e.g., posterior arytenoid, alae nasi, genioglossus). Increases in activity of the dilator muscles of the upper airway decrease the load on the chest wall pumping muscles by decreasing the resistance to inspiratory airflow through the upper airway. Increased activity of these muscles also diminishes the susceptibility of the upper airway to collapse as inspiratory efforts become greater and subpharyngeal pressure becomes more subatmospheric.
In addition, phasic increases in abdominal expiratory muscle electrical activity during expiratory airflow accelerate lung emptying, thereby allowing the time of expiration to decrease. When sufficiently intense, activation of the abdominal muscles reduces end-expiratory lung volume and improves the ability of the diaphragm to generate pressure by favorably affecting its precontraction length, radius of curvature, and alignment with the rib cage. Reductions in end-expiratory lung volume achieved by the expiratory muscles also allow elastic work to be stored in the passive recoil of the chest wall and released suddenly at the onset of inspiration. Sudden release of the recoil pressure of the chest wall thereby “assists” the inspiratory muscles by contributing to the driving pressure to inspiratory airflow. A portion of the inspiratory load is thus assumed by the expiratory muscles.
Finally, hyperinflated, dyspneic patients with COPD often assume a stereotypic body posture that improves diaphragm, neck accessory, and pectoral girdle muscle mechanical advantage. This posture is forward flexion of the trunk, extension of the head and neck, bracing of the pectoral girdle by rounding of the shoulders, and grasping of the thighs with the arms. The effect of this posture is to increase abdominal pressure (thus increasing diaphragm precontraction length and radius of curvature); provide more favorable alignment of the scalenes and sternomastoid with the upper rib cage; and anchor the pectoral girdle muscles, allowing them to apply an inspiratory action on the rib cage. In this posture, transdiaphragmatic pressure is increased and diaphragm and sternomastoid muscle EMG activity is decreased. Patients with advanced COPD also spontaneously adopt pursed-lip breathing to slow expiratory airflow, thus minimizing dynamic airway compression.
Effects of Sleep
Responses to chemical stimuli to breathing are powerfully influenced by CNS state (e.g., sleep vs. waking).16,29,30 Slow-wave and REM sleep depress O2 and CO2 chemosensitivity, with greatest depression occurring in REM sleep. While the subject is awake, apnea does not occur in the presence of even marked hypocapnia, and ventilation is largely independent of changes in PCO2. Rather, ventilation persists even when PaCO2 is less than about 30 to 35 mm Hg. Persistence of ventilation in the setting of hypocapnia (the so-called wakefulness drive to breathe) probably represents the effects on medullary neurons of inputs activated by auditory, visual, and tactile stimuli. In contrast, in the sleeping or anesthetized state, the ventilatory response to CO2 extrapolates to zero ventilation in the hypocapnic range. In fact, apnea occurs when PCO2 falls only 4 to 6 mm Hg below waking eucapnic levels. Sleep-related changes in chemosensitivity, therefore, underlie the recurrent periods of apnea and hyperpnea and exaggerated hypercapnia that occur in some patients with diseases of the lung and chest wall.
The increase in respiratory motor activity induced by derangements in respiratory mechanics is also state dependent; that is, heightened activity in awake subjects is absent in sleeping or anesthetized subjects. REM sleep, in particular, impairs the “load” response and causes collapse of the upper rib cage during inspiration, which adversely affects the level of ventilation as well as its distribution. Descending inhibitory drives to spinal α– and spindle γ–motor neurons in REM sleep cause atonia of all the respiratory muscles except the diaphragm. Muscle spindle γ–efferent activity determines spindle sensitivity by progressively contracting the intrafusal fiber. Accordingly, reductions in muscle spindle γ–efferent activity diminish spindle organ sensitivity and interfere with a mechanism for augmenting respiratory muscle spinal α–motor neuron activity. The diminished or absent load response during sleep and anesthesia probably explains the exaggerated increases in PaCO2 that occur during these periods in patients with lung and chest wall disease. In fact, REM sleep is the period in which PaCO2 is highest and PaO2 lowest in patients with stable COPD (Fig. 143-5).31
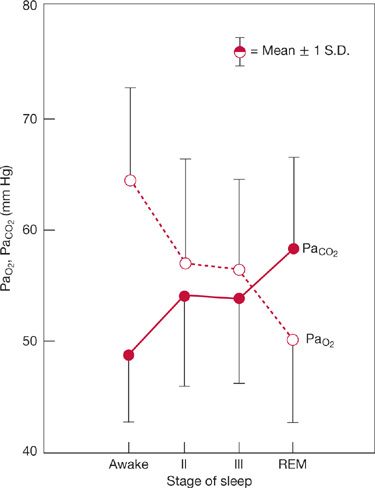
Figure 143-5 Changes in steady-state arterial PCO2 during sleep in eight patients with stable COPD. Note that arterial PCO2 increases and arterial PO2 decreases during sleep. Greatest changes occur during REM sleep. For PaCO2, average increase is 10 mm Hg. (Data from Koo KW, Sax DS, Snider GL. Arterial blood gases and pH during sleep in chronic obstructive pulmonary disease. Am J Med. 1975;58:663–670.)
 CHANGES IN RESPIRATORY STRUCTURE
CHANGES IN RESPIRATORY STRUCTURE
Alterations in respiratory muscles and chest wall anatomy are important to consider in development of hypercapnic respiratory failure.
Respiratory Muscles
The respiratory muscles are highly plastic and undergo changes in structure, biochemistry, and contractile properties in response to chronic increases in load or changes in precontraction length.32–38 Chronic increases in inspiratory muscle activity enhance their strength and endurance.37,38 In animal models, chronic increases in inspiratory load produced by emphysema or inspiratory resistive loading increase diaphragm endurance and the content of oxidant enzymes (e.g., succinate dehydrogenase and citrate synthase) essential for high-energy phosphate synthesis. In patients with COPD, the percentage of slow twitch, fatigue-resistant muscle fibers in the diaphragm and parasternal intercostals is increased.39,40 In chronic asthma, inspiratory and expiratory muscle endurance assessed from the time course of the fall in maximum static pressure is about 40% greater than in normal controls. The effect of COPD per se on inspiratory muscle endurance has not been assessed. In subjects with COPD, however, daily training with inspiratory resistive ventilatory loads increases inspiratory muscle strength by about 40% as reflected by maximum static inspiratory pressure (PImax) over an 8- to 10-week period.
Hyperinflation impairs the force- and pressure-generating ability of the inspiratory muscles by decreasing muscle precontraction length and unfavorably changing muscle alignment with the chest wall.5,27,41,42 In particular, severe hyperinflation alters diaphragm shape (i.e., flattening) and decreases the zone of apposition with the rib cage. Flattening of the diaphragm displaces the vector of contraction force from a rostral–caudal direction to a medial–lateral direction and diminishes the ability of the diaphragm to increase abdominal pressure. Reductions in the zone of apposition diminish the inflationary effects on the lower rib cage produced by increases in abdominal pressure induced by the diaphragm. In extreme cases of hyperinflation, the diaphragm may exert an expiratory action on the lower rib cage and retract the lower rib cage on inspiration (Hoover sign).
In part, hyperinflation-induced impairment in the action of the diaphragm is compensated for by adaptive changes in the intrinsic muscle length–tension characteristic. In emphysematous animals, the active and passive length–tension curve of the costal diaphragm is displaced toward shorter lengths, thereby allowing maximum tension to be developed at significantly shorter lengths and higher lung volumes (Fig. 143-6). The shift in the length–tension curve appears to be the reverse of normal growth, in which muscle length is increased by addition of sarcomeres in series. A similar adaptation in the diaphragm seems to occur in chronically hyperinflated, stable outpatients with COPD.43,44
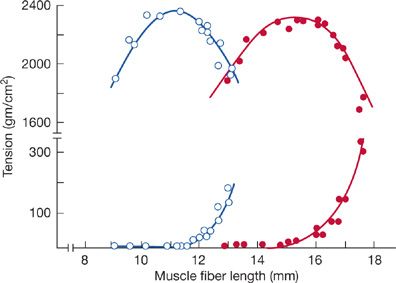
Figure 143-6 Active (upper trace) and passive (lower trace) length–tension (L-T) relationship of costal diaphragm of emphysematous (open circles) and normal hamsters (solid circles), assessed in vitro during electrical stimulation. Note that in emphysematous animals, the L-T curve is displaced toward shorter fiber lengths. This adaptive change in emphysematous animals allows the diaphragm to generate maximal tension (force) at shorter fiber lengths and helps preserve diaphragm contractile performance in the face of considerable hyperinflation. (Reproduced with permission from Supinski GS, Kelsen SG. Effect of elastase-induced emphysema on the force-generating ability of the diaphragm. J Clin Invest. 1982;70(5):978–988.)
Chest Wall Anatomy
Chronic hyperinflation elicits adaptive changes in the pressure–volume (P-V) characteristic of the passive chest wall.36 In animal models of emphysema, the static deflation, chest wall P-V curve is shifted up and to the left, so there is a decrease in elastic recoil at any given lung volume. Shifts in the passive P-V curve are accomplished by a structural remodeling of the rigid structures in the chest wall. The length of the sternum and the lengths of the ribs in anteroposterior and transverse dimensions are increased. This displacement of the chest wall P-V curve diminishes the inspiratory elastic work of breathing during hyperinflation and preserves the zone of apposition of the diaphragm. An increase in the zone of apposition of the diaphragm in hyperinflation preserves the appositional force exerted by the diaphragm on the lower rib cage by virtue of changes in abdominal pressure. If present in patients with COPD, the process is reversible, since recent observations of the thorax after volume reduction surgery or lung transplantation for COPD indicate that the shape of the chest wall can quickly revert to normal.
DECOMPENSATING/MALADAPTIVE RESPONSES
Important maladaptive responses must be considered in the setting of hypercapnic respiratory failure.
 RESPIRATORY MUSCLE FATIGUE
RESPIRATORY MUSCLE FATIGUE
The pathogenesis and clinical manifestations of respiratory muscle fatigue are considered below.
Overview/Definition
Studies in the laboratory and in the clinic indicate that the respiratory skeletal muscles, like muscles in the limbs, fatigue under conditions of intense activity, lead to respiratory failure.45–50 Conditions that increase the level of phasic inspiratory muscle activity, or the duty cycle of breathing, or that decrease the maximal pressure-generating capacity of the muscle, make fatigue more likely. For example, derangements in the mechanical properties of the lung or chest wall or increases in ventilatory drive increase inspiratory muscle contractile activity. Of note, increases in ventilatory drive increase both the peak inspiratory pressure and TI/TT ratio, the latter by causing greater reductions in the duration of expiration than in that of inspiration.
Decreases in inspiratory muscle strength caused by aging, protein–calorie malnutrition, or electrolyte imbalances predispose to fatigue at any given level of inspiratory impedance or ventilation by decreasing PImax. Finally, on the basis of data from animal models, reductions in diaphragm blood flow are likely to decrease the level of muscle activity that leads to fatigue.50,51
Respiratory muscle fatigue has been defined as a loss in muscle capacity to develop force or shorten, resulting from muscle fiber activity under load; which is reversible by rest. In contrast, respiratory muscle weakness has been defined as impairment in the capacity of a fully rested muscle to generate force.
Fatigue is viewed as developing when the muscle is highly active and generating appreciable levels of force. Recovery from fatigue is generally observed over a short time (e.g., minutes to hours). On the other hand, muscle weakness is commonly caused by muscle fiber atrophy, metabolic derangements that impair the ability of actomyosin crossbridges to generate force (e.g., acidosis or electrolyte abnormalities that affect intracellular calcium flux), or chronic reductions in muscle precontraction length that impose a mechanical disadvantage (e.g., hyperinflation of the thorax and its effects on the inspiratory muscles). Implied in the definition of weakness is the idea that alterations in inspiratory muscle function are secondary to alterations in muscle structure or lung volume and hence induce changes in muscle function that are more slowly reversible than fatigue (e.g., days to weeks). In the clinical setting, however, the distinction between muscle weakness and fatigue is difficult and not easily accomplished. Moreover, a close association exists between respiratory muscle weakness and respiratory muscle fatigue. In fact, weak muscles are predisposed to fatigue (see below Pathogenesis of Respiratory Muscle Fatigue).
Fatigue produces complex effects on muscle mechanical output. Fatigue prolongs contraction and relaxation time and depresses the force generated at a given stimulus frequency and fiber length, and reduces the velocity of shortening against a given load.
Depending on the cause of the fatigue, depression of force output can occur at primarily subtetanizing frequencies of muscle stimulation (e.g., <15 to 20 Hz), a condition called low-frequency fatigue, or at frequencies above 50 Hz, a condition called high-frequency fatigue (Table 143-1, Fig. 143-7).47,52 The biochemical and biophysical processes that underlie low-frequency and high-frequency fatigue differ. Muscle force responses to tetanizing frequencies of stimulation (i.e., >50 Hz) are primarily determined by the processes of neuromuscular transmission and muscle excitation. In contrast, muscle mechanical output at subtetanizing frequencies is determined primarily by the processes of excitation–contraction coupling (e.g., calcium release from the sarcoplasmic reticulum, calcium–troponin interactions), perhaps caused, in part, by O2 free radical–induced injury.



