25

Pulmonary Vascular Disease
The lungs are unusual with respect to their blood supply because they possess a double circulation. Like other organs, they receive a share of systemic arterial blood through the bronchial arteries; however, the contribution of the bronchial circulation to the total blood supply of the lungs is modest because only ~1% of the left ventricular output passes into the bronchial arteries. In humans, the bronchial arteries supply blood to the walls of the bronchi and larger blood vessels, the interstitial connective tissue including the interlobular fibrous septa, the hilar lymph nodes, and the visceral pleura. The bronchial circulation is part of the systemic circulation, so the bronchial arteries and veins are similar in structure to other systemic arteries and veins. The major source of blood to the lungs is the pulmonary circulation, which carries the entire output of the right ventricle. The pulmonary circulation is morphologically and functionally unique. Not only does the lung have a more extensive and elaborate circulation than any other organ, but the pulmonary blood vessels maintain a delicate balance of pressure and distribution of flow that facilitates optimum gas exchange. The structure of blood vessels depends to a large extent on the pressure of blood within them. In normal adults, the pulmonary circulation is a low-pressure, low-resistance system. The pulmonary arterial pressure is normally about one sixth of the systemic arterial pressure. Accordingly, the pulmonary blood vessels that are faced with lower pressures than the systemic vessels have thinner walls and wider lumina than their systemic counterparts. Being initially thinner and less muscular, pulmonary blood vessels react in a striking fashion to an elevated intravascular pressure.
Fixation and Staining Methods for Examination of the Pulmonary Vasculature
It is desirable that surgical lung biopsy specimens and lungs at autopsy be fixed in a state of distention. Otherwise, collapse of lung tissue may lead to crenation of the elastic laminae and induce a state of spurious medial hypertrophy in muscular pulmonary arteries. Surgical lung biopsy specimens are most conveniently fixed by injecting them through multiple punctures in the pleura with a 25-gauge butterfly needle connected to a 5 mL syringe. This technique induces slight edema of the interlobular fibrous septa, which does not lead to difficulties in interpretation and is a small price to pay for the excellent display of pulmonary microanatomy.
Lungs at autopsy may be distended by endobronchial instillation of an appropriate fixative, usually formalin. The technique of postmortem angiography followed by histology in the assessment of pulmonary vascular diseases has been advocated by some workers1,2 but criticized by others.3–5 A pressure of 100 cm H20 or 75 mm Hg is used to force a barium-gelatin mixture into the pulmonary arteries. Vessels containing barium are then counted in histologic sections. This technique has been purported to show a reduction in the number of pulmonary arteries in various types of pulmonary arterial hypertension.2,6 In contrast, direct counts of pulmonary blood vessels have failed to demonstrate a reduction in the number of pulmonary arteries.4,7,8 Barium-gelatin mixture is erratic in its penetration of the pulmonary vasculature. Postmortem pulmonary angiography cannot distinguish between absence of blood vessels and total or subtotal occlusion by constriction or intimal thickening.
The walls of blood vessels are composed essentially of collagen, smooth muscle, and elastic tissue. Using the conventional hematoxylin and eosin (H&E) staining method, all these tissues appear pink and cannot be distinguished from one another. It is necessary to use a connective tissue stain such as the Miller elastic–van Gieson method for evaluating the pulmonary vasculature.9 Representative sections should also be stained by the Perls Prussian blue method for ferric iron to detect hemosiderosis, which is one of the features of chronic pulmonary venous hypertension.
Pulmonary Hypertension
Definition
In adults, the normal pulmonary artery pressure at rest is 16/7 mm Hg, with a mean of 12 mm Hg.10 The criterion used by the National Heart, Lung, and Blood Institute Registry of Primary Pulmonary Hypertension was a mean pulmonary artery pressure greater than 25 mm Hg at rest, or 30 mm Hg with exercise at cardiac catheterization.11
Nomenclature and Classification
Pulmonary hypertension is a common condition that has many causes and associations, and has been the subject of many attempts at classification. In the majority of patients, clinical examination together with relevant investigations reveals a cause such as underlying heart or lung disease. These cases have been classified as secondary pulmonary hypertension. In a small minority of patients, the cause of the pulmonary hypertension remains unexplained despite exclusion of predisposing lesions. In 1975, a committee of the World Health Organization (WHO) suggested that such cases should be designated primary pulmonary hypertension.12 Unfortunately, many authors have used this term to refer specifically to patients with pulmonary hypertension of unknown cause in whom histologic examination disclosed plexiform lesions, so that in practice it has had two different meanings.3 The term unexplained pulmonary hypertension was then introduced to refer to cases of pulmonary hypertension in which clinical examination and investigation revealed no cause.13 Pulmonary hypertension has also been classified as hyperkinetic, passive, obstructive, and vasoconstrictive according to the dominant pathophysiologic mechanism involved.10 A new diagnostic classification of pulmonary hypertension was proposed at a WHO meeting in 1998 (Table 25–1). This classification recognizes that the tissue pathology of pulmonary hypertension caused by different agents may be similar, and that the treatment may be identical.14
1. Pulmonary arterial hypertension 1.1 Primary pulmonary hypertension (a) Sporadic (b) Familial 1.2 Related to: (a) Collagen vascular disease (b) Congenital systemic-to-pulmonary shunts (c) Portal hypertension (d) HIV infection (e) Drugs/toxins (1) Anorexigens (2) Other (f) Persistent pulmonary hypertension of the newborn (g) Other 2. Pulmonary venous hypertension 2.1 Left-sided atrial or ventricular heart disease 2.2 Left-sided valvular heart disease 2.3 Extrinsic compression of central pulmonary veins (a) Fibrosing mediastinitis (b) Adenopathy/tumors 2.4 Pulmonary veno-occlusive disease 2.5 Other 3. Pulmonary hypertension associated with disorders of the respiratory system and/or hypoxemia 3.1 Chronic obstructive pulmonary disease 3.2 Interstitial lung disease 3.3 Sleep disordered breathing 3.4 Alveolar hypoventilation disorders 3.5 Chronic exposure to high altitude 3.6 Neonatal lung disease 3.7 Alveolar-capillary dysplasia 3.8 Other 4. Pulmonary hypertension due to chronic thrombotic and/or embolic disease 4.1 Thromboembolic obstruction of proximal pulmonary arteries 4.2 Obstruction of distal pulmonary arteries (a) Pulmonary embolism (thrombus, tumor, ova, and/or parasites, foreign material) (b) In situ thrombosis (c) Sickle cell disease 5. Pulmonary hypertension due to disorders directly affecting the pulmonary vasculature 5.1 Inflammatory (a) Schistosomiasis (b) Sarcoidosis (c) Other 5.2 Pulmonary capillary hemangiomatosis |
HIV, human immunodeficiency virus.
Causes
The pathophysiological mechanisms that may result in pulmonary hypertension are as follows:
• Increased pulmonary blood flow (hyperkinetic pulmonary hypertension)
• Prolonged left atrial hypertension (passive pulmonary hypertension)
• Organic vascular obstruction (obstructive pulmonary hypertension)
• Pulmonary vasoconstriction (vasoconstrictive pulmonary hypertension)
Pulmonary hypertension is associated with many diseases, and it is important to realize that pulmonary hypertension arising in a given disease process may utilize more than one of the four pathophysiological mechanisms listed above. For example, pulmonary hypertension complicating usual interstitial pneumonia may have obstructive and vasoconstrictive components.
Pretricuspid shunts |
Atrial septal defects |
Anomalous pulmonary venous drainage |
Post-tricuspid shunts |
Ventricular septal defect |
Persistent truncus arteriosus |
Single ventricle |
Eisenmenger’s complex |
Patent ductus arteriosus |
Aortopulmonary septal defect |
Complete atrioventricular canal defect |
Rupture of aortic aneurysm into pulmonary artery |
Rupture of ventricular septum after myocardial infarction |
Postoperative systemic-pulmonary shunt procedure |
Increased Pulmonary Blood Flow
Shunts between the systemic and pulmonary circulations increase pulmonary blood flow (Table 25–2). Pulmonary hypertension may develop if the shunt is large enough and if there is no concomitant pulmonary stenosis to protect the pulmonary blood vessels. It is useful to consider a shunt as being either pretricuspid or post-tricuspid because the risk to the pulmonary circulation is different in each case.
Pretricuspid Shunts
Pretricuspid shunts occur at the level of the atria. The increase in pulmonary blood flow is due to blood entering the systemic venous stream before it arrives in the right ventricle. Congenital heart diseases accompanied by this hemodynamic abnormality include high atrial septal defects such as those of secundum or sinus venosus type, and anomalous pulmonary venous drainage, without obstruction, into the right atrium or systemic or portal veins.
Post-Tricuspid Shunts
A large communication between the left ventricle or aorta and the right ventricle or pulmonary arteries leads to elevation of the pulmonary artery pressure due to an increase in pulmonary blood flow and direct transmission of systemic pressure into the pulmonary circulation. Post-tricuspid shunts differ in their natural history from pretricuspid shunts in that pulmonary hypertension is usually present from birth and not acquired in later life. Most of the diseases with post-tricuspid shunts are congenital anomalies of the heart and great vessels (Table 25–2) such as complete atrioventricular canal defect, ventricular septal defect, single ventricle, Eisenmenger’s complex, patent ductus arteriosus, aortopulmonary septal defect, cardioaortic fistula, and persistent truncus arteriosus. Rarely, a post-tricuspid shunt may be acquired. Examples are rupture of an aortic aneurysm into the pulmonary artery and rupture of the interventricular septum following myocardial infarction. Pulmonary hypertension may follow the surgical creation of a post-tricuspid shunt between a systemic artery and a pulmonary artery during the palliative treatment of cyanotic congenital heart disease with diminished pulmonary blood flow.
Prolonged Left Atrial Hypertension
An increase in the left atrial pressure may be transmitted back through the pulmonary capillaries to elevate the pulmonary artery pressure and give rise to passive pulmonary hypertension. However, an increase in the left atrial pressure of more than 18 mm Hg causes reflex vasoconstriction in the pulmonary arteries and a disproportionate rise in the pulmonary artery pressure.10 Most causes of left atrial hypertension are acquired and include mitral stenosis, mitral incompetence, left atrial myxoma, and any cause of chronic left ventricular failure including ischemic heart disease, systemic hypertension, aortic stenosis, aortic incompetence, myocarditis, and cardiomyopathy. Congenital mitral stenosis may cause prolonged left atrial hypertension.
Organic Vascular Obstruction
A significant reduction in the total cross-sectional area of the pulmonary vascular bed increases the resistance to blood flow and so causes pulmonary hypertension. However, the pulmonary vascular reserve is enormous. It is only after ablation of 60% of the lung that pulmonary artery pressure rises.10 Life may still exist after removal of 80% of the lungs. Pulmonary embolism is the commonest cause of obstructive pulmonary hypertension. Other acquired lesions that may cause obstructive pulmonary hypertension include pulmonary fibrosis, fibrosing mediastinitis, pulmonary veno-occlusive disease, and pulmonary capillary hemangiomatosis. Congenital lesions that may cause obstructive pulmonary hypertension include multiple stenoses of the pulmonary arteries and congenital stenosis of the pulmonary veins. In total anomalous pulmonary venous drainage, there is a high pulmonary blood flow, and this leads to pulmonary hypertension. However, pulmonary hypertension in this condition may also be related to pulmonary venous obstruction due to kinking of the long anomalous veins or to stenotic lesions in them, or by pressure of contiguous structures on them.
Pulmonary Vasoconstriction
Because of the sparsity of smooth muscle in vessel walls, vasomotor activity in pulmonary blood vessels is slight in contrast to that of the much more muscular systemic vessels. Pulmonary arteries differ from systemic arteries in that they constrict rather than dilate in response to hypoxia, and persistent alveolar hypoxia induces pulmonary hypertension.15 The causes of chronic hypoxic pulmonary hypertension include upper airway obstruction due to enlarged adenoids and malformations of the facial skeleton; chronic lung diseases such as chronic airflow obstruction, bronchiectasis, cystic fibrosis, lung fibrosis and bronchial asthma; alveolar hypoventilation in the obesity-hypoventilation (pickwickian) syndrome, sleep apnea syndrome and neuromuscular disorders, and disorders of the thoracic skeleton including kyphoscoliosis and ankylosing spondylitis. Millions of people live at high altitude where the diminished barometric pressure leads to a decreased partial pressure of oxygen in the alveoli. Many of these native highlanders are chronically hypoxic and have mild pulmonary hypertension, although they are healthy. Only a few develop chronic mountain sickness (Monge’s disease) when the pulmonary hypertension becomes more severe.
Cor Pulmonale
An expert committee of the WHO defined cor pulmonale as “hypertrophy of the right ventricle resulting from diseases affecting the function and/or the structure of the lung, except where these alterations are the result of diseases that primarily affect the left side of the heart or of congenital heart disease.”16 Right ventricular hypertrophy may be assessed morphologically, electrocardiographically, or radiologically. Unfortunately, in spite of this precise definition, cor pulmonale continues to be an ambiguous term because it is frequently applied to patients with right ventricular failure in whom there is no evidence of right ventricular hypertrophy.
Assessment of Right Ventricular Hypertrophy
Assessment of right ventricular hypertrophy is an important part of the autopsy in a subject with airway, interstitial, or vascular disease of the lungs, particularly if there were no measurements of pulmonary artery pressure during life (Fig. 25–1). Total heart weight17 and right ventricular wall thickness18 correlate poorly with right ventricular mass. Measurement of thickness of the right ventricular wall at autopsy is notoriously unreliable. An oblique cut may give a spurious impression of right ventricular hypertrophy. Conversely, right ventricular hypertrophy may be missed if there is coexistent dilatation that diminishes the thickness of the wall. A 100-g right ventricle may measure from 3 to 11 mm in thickness, whereas a right ventricle measuring 5 mm in thickness may weigh from 30 to 120 g.18 The method of Fulton et al19 is the most satisfactory for evaluating ventricular hypertrophy at autopsy.17 They removed the epicardial fat, detached the atria at the atrioventricular groove, and then determined the weight of the free wall of the right ventricle and the weight of the left ventricle and interventricular septum. Right ventricular hypertrophy is considered to be present when the free wall of the right ventricle weighs 80 g or more. Left ventricular hypertrophy is considered to be present when the weight of the left ventricle and interventricular septum exceeds 225 g. The ratio of the weight of the left ventricle and interventricular septum to that of the right ventricle normally lies between 2.3 and 3.3. In isolated right ventricular hypertrophy, the ratio is always less than 2. The fetus has a high pulmonary vascular resistance and the ratio of left to right ventricular weight is normally 1.5.20 The adult ratio is achieved 10 months after birth.21
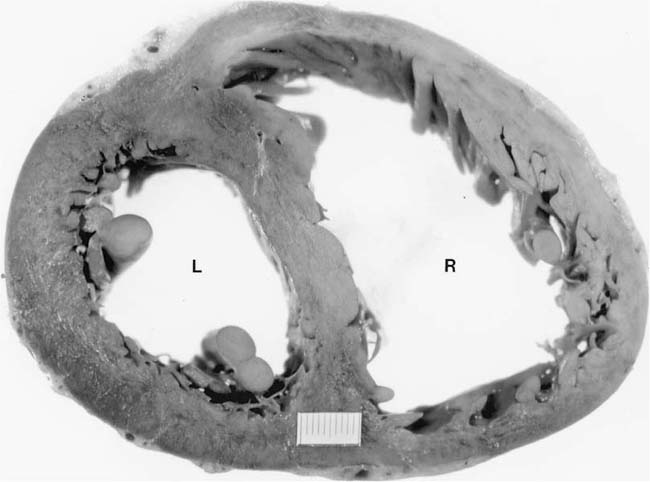
FIGURE 25–1 Transverse section through the heart of a 25-year-old woman with systemic sclerosis and severe pulmonary hypertension (pulmonary artery mean pressure 61 mm Hg). There was hypertrophy of the right ventricle (R), which was somewhat masked by dilatation. The right ventricular free wall ranged from 0.5 to 0.8 cm in thickness. The free wall of the right ventricle weighed 120 g (upper limit of normal 80 g). The left ventricle (L) and septum weighed 150 g (upper limit of normal 225 g). The total heart weight was 360 g. The lungs were devoid of fibrosis but showed pulmonary arteriopathy with plexiform lesions. (Courtesy of Dr. H. A. Heggtveit.)
Hypertensive Pulmonary Vascular Disease
The structural changes that may occur in response to altered hemodynamics are much greater in the pulmonary circulation than in the systemic vasculature. In contrast to the systemic circulation, the reaction of the pulmonary vasculature to increased blood pressure is not uniform. The reaction varies with not only the degree of pulmonary hypertension but also the cause. Different causes of pulmonary hypertension produce different patterns of hypertensive pulmonary vascular disease (Table 25–3).3 Within the pulmonary arterial system, lesions may be found in the pulmonary trunk and main pulmonary arteries. However, these changes are never so severe and so varied as those encountered in the muscular pulmonary arteries and pulmonary arterioles, which comprise the most reactive parts of the pulmonary vasculature. Pulmonary hypertension differs from systemic hypertension in that in certain diseases, characteristic changes also occur in the pulmonary capillaries and veins. There is considerable variation in pulmonary vascular reactivity from individual to individual. This means that the same cause of pulmonary hypertension may be present in various subjects to the same extent, but the degree of pulmonary hypertension and the severity of the hypertensive pulmonary vascular disease may vary widely. The wide range of pulmonary vascular lesions that may be encountered in patients with pulmonary hypertension is illustrated in the following discussion.
Pattern | Causes |
|---|---|
Pulmonary arteriopathy with plexiform lesions | Systemic-to-pulmonary shunts, primary pulmonary arterial hypertension, portal hypertension/liver disease, HIV infection, anorexigenic drugs |
Congestive pulmonary vasculopathy | Left ventricular failure, mitral valve disease, left atrial myxoma, cor triatriatum, stenosis or compression of main pulmonary veins, pulmonary veno-occlusive disease |
Thrombotic pulmonary arteriopathy | Thromboembolism, thrombotic obstruction of distal pulmonary arteries, diminished pulmonary blood flow |
Hypoxic pulmonary arteriopathy | Upper airway obstruction, chronic Airflow obstruction, alveolar hypoventilation syndromes, thoracic skeletal disorders, residence at high altitude |
Systemic-to-Pulmonary Shunts
Pretricuspid shunts occur at the level of the atria and include high atrial septal defects such as those of the secundum or sinus venosus type. Regardless of the anatomic type, the physiologic consequences of atrial septal defects are the result of shunting of blood from one atrium to the other. The direction and magnitude of the shunt are determined by the size of the defect, and the relative distensibility of the two ventricles.22 A small defect (<0.5 cm diameter) is associated with a small shunt and no hemodynamic sequelae. Fifty-seven percent of small atrial septal defects close spontaneously at a mean age of 8.4 years.23 A sizable defect (>2 cm diameter) may be associated with a large shunt and significant hemodynamic consequences. At birth the thickness of the ventricles is similar, and their distensibilities do not differ significantly. Therefore, there is little or no shunting of blood through an atrial septal defect. However, once the right ventricular pressure falls, the wall of the right ventricle becomes thinner relative to the left ventricle, whereas its distensibility becomes relatively greater. As a result, a left-to-right shunt develops through the atrial septal defect, thereby increasing the pulmonary blood flow. In young people with atrial septal defects and pretricuspid shunts the pulmonary blood flow may be three times greater than normal but the pulmonary vascular resistance is not elevated because of the enormous distensibility of the pulmonary vasculature. However, an increase in pulmonary blood flow of this magnitude for many years eventually leads to the development of pulmonary hypertension in ~30% of subjects.10 Hypertensive pulmonary vascular disease is usually delayed until the fourth decade,24 only rarely occurring in childhood25 or early adult life.
In post-tricuspid shunts, such as a ventricular septal defect, the magnitude of the shunt is proportional to the area of the defect and to the difference in pressure between the two ventricles. Associated stenosis of the pulmonary valve increases the resistance to outflow from the right ventricle and diminishes the magnitude of the left-to-right shunt. Conversely, stenosis of the aortic valve elevates left ventricular pressure and increases the shunt of blood into the right ventricle. At birth there is normally a rapid fall in pulmonary vascular resistance as the ductus arteriosus closes and the pulmonary arteries dilate, but in the presence of a large ventricular septal defect, this fall in pulmonary vascular resistance and pressure may be delayed for several weeks. Only when the resistance begins to decline does the left-to-right shunt increase. Thus, affected infants do not usually develop symptoms during the first 4 weeks of life. However, as the shunt subsequently increases in magnitude, the child may develop pulmonary edema. This is partly the consequence of the increased pulmonary blood flow leading to a transudate of fluid through the dilated pulmonary vasculature, and partly the effect of left ventricular failure. Ventricular septal defect is the most common congenital cardiac abnormality in infants and children, and its natural history depends on its size. Most defects are small. Seventy-five percent of small defects, and 10% of large defects close spontaneously; 25 to 40% of defects that close spontaneously do so by the time the child is 2 years old; 90% of those that eventually close do so by the time the child is 10 years old.22 Thus severe pulmonary hypertension occurs in only a small proportion of patients with ventricular septal defect.
Pulmonary hypertension with advanced hypertensive pulmonary vascular disease is sometimes encountered in young children with transposition of the great arteries with an intact ventricular septum.26 The cause of the pulmonary hypertension is unexplained. Even though there is a patent foramen ovale or atrial septal defect in these cases, advanced hypertensive pulmonary vascular disease would not be expected at such an early age. It has been suggested that the pulmonary arteries in such patients have structural defects that predispose to the development of severe pulmonary vascular lesions.3,26,27

FIGURE 25–2 Transverse section of part of the media of the pulmonary trunk from a 54-year-old man with chronic obstructive pulmonary disease and severe right ventricular hypertrophy. The free wall of the right ventricle weighed 204 g (upper limit of normal 80 g). Most of the elastic fibers are short and irregular. This is the adult elastic tissue configuration seen in acquired pulmonary hypertension [elastic–van Gieson (EVG), ×500].
Elastic Pulmonary Arteries
The changes that normally occur in the elastic tissue configuration of the media of the pulmonary trunk after birth are described in Chapter 1. In general, there is progressive fragmentation and reduction in density of elastic fibers associated with a relative reduction in medial thickness. When pulmonary hypertension complicates a pretricuspid shunt such as an atrial septal defect, pulmonary hypertension develops after the transition from an aortic to an adult configuration of elastic tissue in the pulmonary trunk. Accordingly, an adult elastic tissue pattern is indicative of acquired pulmonary hypertension (Fig. 25–2). On the other hand, in a post-tricuspid shunt such as a ventricular septal defect, the pulmonary hypertension is present from birth and the elastic tissue in the pulmonary trunk fails to fragment. Thus, an aortic configuration of elastic tissue is indicative of pulmonary hypertension present from infancy (Fig. 25–3).28 The intrapulmonary elastic arteries show nonspecific thickening of the tunica media due to an increase in smooth muscle cells and elastic fibers. The larger proximal elastic pulmonary arteries may show atheromatous changes whereas the peripheral ones may exhibit mild intimal fibrosis.29
Muscular Pulmonary Arteries and Arterioles
Progressive changes develop in the intima and media of muscular pulmonary arteries and pulmonary arterioles in patients with large systemic-to-pulmonary shunts (Table 25–4). The lesions tend to follow a sequence irrespective of the precise nature of the defect causing the shunt, but related to a progressive increase in pulmonary artery pressure and pulmonary vascular resistance, and to a reduction in pulmonary blood flow. The earliest structural change is medial hypertrophy of muscular pulmonary arteries accompanied by muscularization of pulmonary arterioles. In more severe cases, this is complicated by a cellular intimal proliferation, which may be followed by concentric laminar intimal fibrosis. The most advanced changes comprise plexiform lesions, dilatation lesions, fibrinoid necrosis, and arteritis.

FIGURE 25–3 Transverse section of part of the media of the pulmonary trunk from a 2-year-old girl with Down syndrome complicated by a ventricular septal defect and patent ductus arteriosus. The elastic fibers are long, broad, and parallel. They form a series of laminae such as occur in the aorta. This is the fetal, aortic elastic tissue configuration seen in neonates and in cases of pulmonary hypertension present from birth (EVG, ×500).
Pulmonary arteries |
Medial hypertrophy |
Muscularization of arterioles |
Cellular intimal proliferation |
Concentric laminar intimal fibrosis |
Plexiform lesions |
Dilatation lesions |
Veinlike branches |
Angiomatoid lesions |
Fibrinoid necrosis and arteritis |
Occasional nonspecific changes |
Tortuosity |
Generalized dilatation |
Longitudinal muscle |
Adventitial thickening |
Thrombi |
Pulmonary veins |
Nonocclusive intimal fibrosis |
Muscularization of media |
Lung parenchyma |
Increased alveolar macrophages |
Intraalveolar hemorrhage |
Hemosiderin-laden macrophages |
Focal intraalveolar fibrosis |
Cholesterol granulomas |
Mast cell hyperplasia |
Increased endocrine cells in terminal bronchioles |
Hypertrophy of bronchiolar smooth muscle |
Peribronchial lymphoid aggregates |
Medial Hypertrophy, Muscularization of Arterioles
There is in vivo and in vitro evidence of phenotypic heterogeneity within the smooth muscle cell population of the arterial media.30 Numerous functions are required of vascular smooth muscle cells under both normal and pathologic conditions. These include contraction, proliferation, and synthesis of extracellular matrix. These different smooth muscle cell functions may require different phenotypes. Normal pulmonary artery smooth muscle cells are not proliferating, migrating, or synthesizing extracellular matrix. Their quiescent state is maintained by the endothelial cells that inhibit proliferation and synthesis of extracellular matrix. In chronic pulmonary hypertension, some medial smooth muscle cells are likely to hypertrophy and retain contractile functions, whereas others are likely to exhibit increased proliferation or synthesis of collagen and elastin. These cellular responses are stimulated by hypoxia, shear stress, blood borne and locally produced mitogens, and growth factors, which activate a cascade of intracellular signaling mechanisms that promote smooth muscle cell growth or the synthesis of extracellular matrix.
The earliest morphologic evidence of hypertension in the muscular pulmonary arteries is thickening of the media (Fig. 25–4). This is usually called medial hypertrophy, although it is occasionally termed medial hyperplasia. Both terms are correct because there is an increase in both the number and size of smooth muscle cells. The degree of medial hypertrophy may vary considerably, but there is a rough correlation between the medial thickness of the muscular pulmonary arteries and the pulmonary artery pressure and calculated resistance.27,31 The average medial thickness of muscular pulmonary arteries not distended postmortem ranges between 3 and 7% of the external diameter in normal adults.32 In mild medial hypertrophy, the average medial thickness lies between 7 and 10%. Moderate medial hypertrophy is present when the average medial thickness ranges between 10 and 15%. In severe medial hypertrophy the average medial thickness exceeds 15%.3 In infants and young children with congenital cardiac shunts, the average medial thickness may attain 25%.
The earliest morphologic sign of pulmonary hypertension is muscularization of arterioles (Fig. 25–5).2 Pulmonary arterioles are defined as arterial vessels measuring less than 100 μm external diameter (see Chapter 1). The transition from the muscular to the nonmuscular part of the pulmonary arterial tree normally takes place in vessels with a diameter of 70 to 100 μm. This transition is not sharp because the media changes gradually from a complete layer of smooth muscle to a discontinuous one. The circularly arranged smooth muscle cells in this intermediate segment follow an ever-widening spiral course so that muscular and nonmuscular portions alternate. Vessels in this intermediate segment are sometimes referred to as partially muscular arteries.1 The distal portions of these pulmonary arterioles, which are nonmuscular, have a wall consisting of a single elastic lamina, which is a continuation of the external elastic lamina. In pulmonary hypertension, the walls of pulmonary arterioles as small as 20 to 30 μm in diameter develop a thick muscular media sandwiched between distinct internal and external elastic laminae. In infants, such muscularized pulmonary arterioles may be even smaller. Ultrastructural studies in lung biopsy material from children with congenital heart defects have shown that the new media arises as a result of hypertrophy, division, and differentiation of precursor smooth muscle cells, pericytes, and intermediate cells, which are normally found in the nonmuscular and partially muscular pulmonary arterioles, respectively.33 There is also synthesis of elastic tissue with the formation of a new internal elastic lamina.
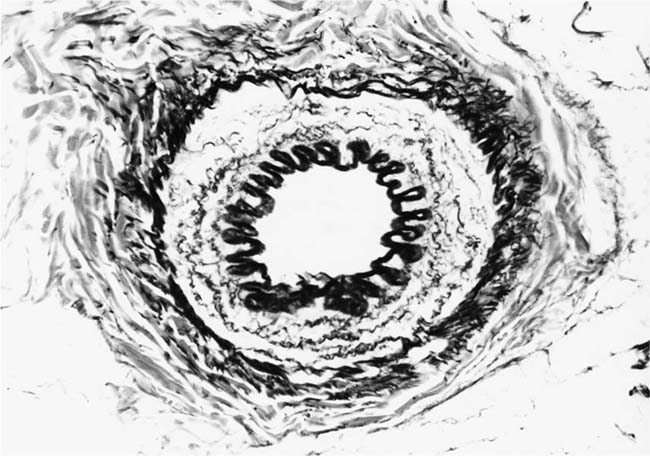
FIGURE 25–4 Muscular pulmonary artery from a 10-month-old girl with a large ventricular septal defect. It shows pronounced medial hypertrophy. The crenation of the internal and external elastic laminae is consistent with some degree of collapse or vasoconstriction (EVG, ×500).
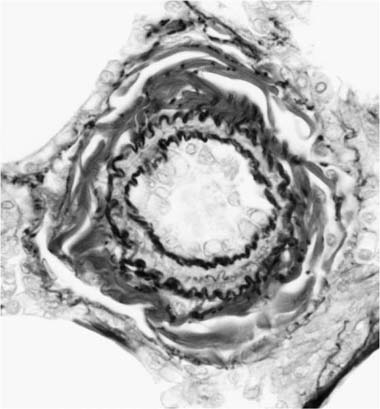
FIGURE 25–5 Pulmonary arteriole from the same case as Fig. 25–4. The vessel is muscularized and has a thick media of circular smooth muscle sandwiched between internal and external elastic laminae (EVG, ×800).
It has been stated that the medial hypertrophy of muscular pulmonary arteries and muscularization of pulmonary arterioles in infants with post-tricuspid shunts is due to retention of the fetal type of pulmonary vasculature rather than to a postnatal increase in vascular smooth muscle. This is unlikely to be the case. In prenatal life the medial thickness of pulmonary arteries in fetuses with ventricular septal defects is the same as that in normal fetuses.34 After birth, the pulmonary arteries dilate in response to the increase in pulmonary blood flow irrespective of whether or not there is a cardiac defect. However, in normal neonatal pulmonary arteries the media becomes thinner over the succeeding weeks and months. If a ventricular septal defect is present, the media increases in thickness after the initial partial thinning. Within a few weeks or months it may be much thicker than it was at the time of birth. This implies that persistence of the fetal type of pulmonary vasculature plays only a limited role, if any, in the development of medial hypertrophy and muscularization of pulmonary arterioles in infants with ventricular septal defects. In pretricuspid congenital cardiac shunts, such as atrial septal defects, which are characterized by a long period of high pulmonary blood flow before the onset of pulmonary hypertension, the pulmonary arteries are initially thin walled and the pulmonary arterioles devoid of a muscular media. As pulmonary hypertension supervenes, there is associated medial hypertrophy of the muscular pulmonary arteries and muscularization of the pulmonary arterioles. Thus, the appearances come to resemble those seen in post-tricuspid shunts.
Cellular Intimal Proliferation
This constitutes the first stage in the development of intimal thickening, and is sometimes the only intimal change observed in infants and young children with systemic to pulmonary shunts.27 Cellular intimal proliferation tends to occur in pulmonary arteries 100 to 150 μm in diameter, close to their origins from parent vessels. It occurs more frequently in arterial branches formed by symmetrical regular dichotomy than in supernumerary arteries.35 A layer of cells forms between the endothelial basement membrane and the internal elastic lamina. The cells have elongated nuclei that initially tend to be arranged perpendicularly to the media. Later, they develop a circumferential pattern (Fig. 25–6). There is ultrastructural36 and immunohistochemical37 evidence that the cellular intimal proliferation is derived from smooth muscle cells from the inner half of the tunica media that migrate into the intima through gaps in the internal elastic lamina. These gaps may be natural, or may arise in pulmonary hypertension as a result of elastase activity.38 The deposition of smooth muscle cells in between the endothelial basement membrane may cause considerable narrowing of muscular pulmonary arteries. In smaller branches, the lumen is often reduced to a barely recognizable slit.
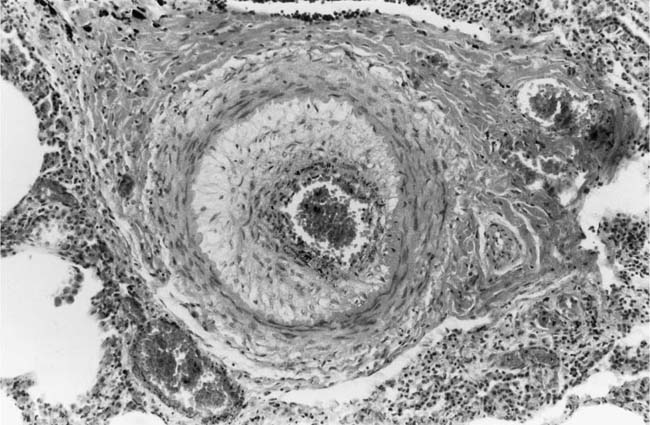
FIGURE 25–6 Muscular pulmonary artery from a 12-year-old girl with cor triloculare biatriatum. Medial hypertrophy is present, and the lumen is narrowed by a cellular intimal proliferation [Hematoxylin and eosin (H&E), ×200]. (Courtesy of Professor D. A. Heath.)
The endothelium in patients with congenital heart disease is thickened and shows ultrastructural changes suggestive of increased metabolic function.38 Although endothelium is not directly responsible for the cellular intimal proliferation, it may regulate the proliferation, migration, and synthetic activity of the smooth muscle cells.30
Concentric Laminar Intimal Fibrosis
Deposition of collagen fibers in between the smooth muscle cells that comprise the cellular intimal proliferation gives rise to concentric laminar fibrosis with an onion-skin appearance (Fig. 25–7). Later, elastic tissue is also deposited as fine concentric fibrils. In its most severe form, concentric laminar intimal fibrosis leads to almost total obliteration of the lumen, which may be associated with atrophy of the tunica media. As with cellular intimal proliferation, concentric laminar intimal fibrosis tends to develop in muscular pulmonary arteries 100 to 150 μm in diameter, close to their origins from parent vessels. It is also more prevalent in peripheral branches formed by symmetrical regular dichotomy than in supernumerary arteries.35 Concentric laminar intimal fibrosis is uncommon below the age of 2 years.
Plexiform Lesions
These lesions, which signify an advanced form of hypertensive pulmonary vascular disease, occur in pulmonary arterial branches measuring between 100 and 150 μm in diameter, shortly after their origin from larger arteries. They are slightly more common in supernumerary arteries than in symmetrical regular dichotomous branches.35 About two thirds of plexiform lesions are located within preacinar pulmonary arteries, and one third are related to intraacinar pulmonary arteries.39 The typical plexi-form lesion has a striking appearance and has two basic elements. The first consists of a circumscribed dilatation of a pulmonary artery branch close to its origin from a parent artery (Fig. 25–8). In this dilated segment, there is destruction of the arterial wall, with pronounced thinning of the media, loss of smooth muscle cells, and disappearance of the internal elastic lamina and sometimes also the external elastic lamina. The second element is a plexus of narrow slitlike channels within the dilated segment (Fig. 25–9). These channels, which are lined by cells with hyperchromatic nuclei, resemble a vascular plexus. The distal part of the plexiform lesion drains into a dilated and thin-walled vessel, the wall of which usually consists of a single elastic lamina (Fig. 25–8). This channel, or its branches, terminates in the alveolar capillaries.40 Reconstructions of plexiform lesions based on serial sections have shown the occasional presence of tiny communications between the dilated distal vessels and branches of other pulmonary arteries, bronchial arteries, and pulmonary veins.35
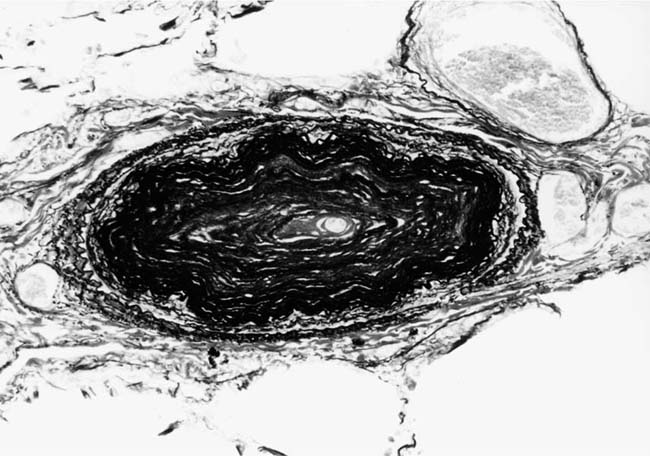
FIGURE 25–7 Muscular pulmonary artery showing severe concentric laminar intimal fibrosis with almost total obliteration of the lumen. From a 25-year-old man with a large aortopulmonary septal defect (EVG, ×200).
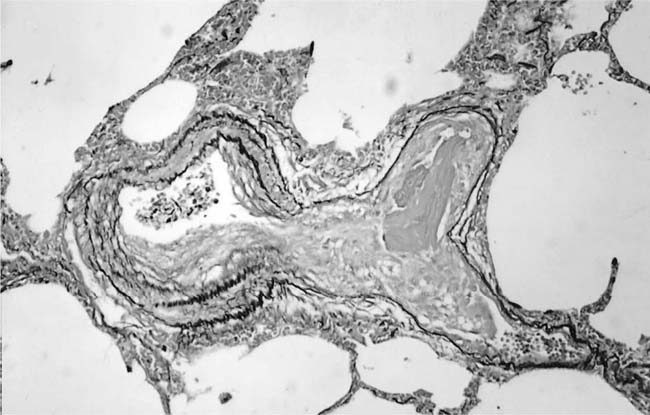
FIGURE 25–8 Plexiform lesion arising in a lateral branch of a muscular pulmonary artery. The parent muscular pulmonary artery (left half of figure) shows medial hypertrophy and intimal fibrosis. The lateral branch (right half of figure) shows aneurysmal dilatation associated with destruction of the internal elastic lamina. The lumen of the lateral branch is occupied by a cellular proliferation and some thrombus. In the lower right corner, the distal part of the plexiform lesion is draining into a dilated thin-walled vessel, which terminates in the alveolar capillaries. From a patient with pulmonary arterial hypertension and a history of ingestion of aminorex (EVG, ×200). (From Kay et al,236 with permission.)
Plexiform lesions vary in structure according to their age. In the early stages of their development, they are cellular, and numerous plump cells line the vascular spaces. The tissue separating the vascular spaces contains rounded cells and small quantities of collagen. The media of the parent artery often contains a focus of fibrinoid necrosis, which may extend into the wall of the sac. The vascular spaces commonly contain thrombi. More advanced plexiform lesions contain large quantities of collagen, which is deposited between the vascular channels and which may encroach on them (Fig. 25–10). The regions of fibrinoid necrosis and thrombosis undergo organization to form masses of collagen. Finally, elastic fibers become deposited. These mature plexiform lesions may appear different from the younger ones but can still be recognized as dilated sacs containing irregular bands of fibroelastic tissue in which a plexus of vascular channels persists (Fig. 25–11). Such mature plexiform lesions can be mistaken for organized thrombi.
Plexiform lesions are observed mainly in adolescents and adults and are uncommon before the age of 2 years, although they have been described in infants as young as 8 weeks of age.41 Their numbers vary greatly. In some cases, numerous plexiform lesions may be seen in a single histologic section, whereas in other cases they are so scarce that multiple sections, sometimes from different blocks, are necessary to find them.
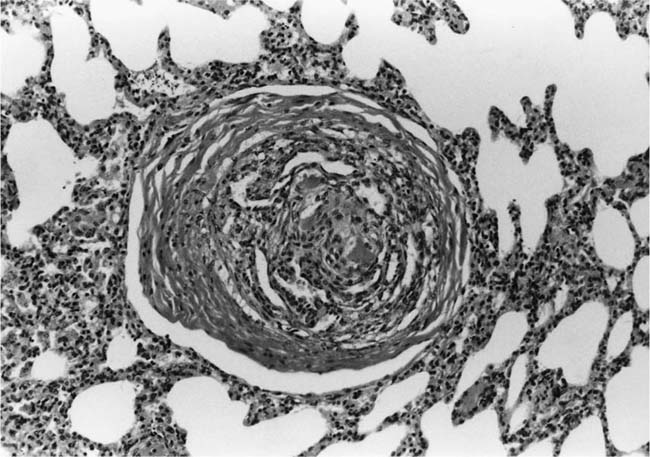
FIGURE 25–9 Transverse section of a muscular pulmonary artery showing a plexiform lesion. In this dilated segment, there is destruction of the arterial wall with pronounced thinning of the media and loss of smooth muscle cells. The lumen is occupied by a plexus of narrow slitlike channels lined by cells with hyperchromatic nuclei. From the patient illustrated in Fig. 25–4 (H&E, ×200).
The pathogenesis of the plexiform lesion is incompletely understood. Some workers consider it to be a proliferative response to vascular injury produced by intense vasoconstriction,42,43 and others believe it to represent an abnormal form of intravascular angiogenesis.44 The specific location of plexiform lesions at points of branching of the muscular pulmonary arteries, and the fact that they do not occur in bronchial arteries,39 suggests that shear stress or turbulence may influence their development.
Ultrastructural studies45 and experimental studies of pulmonary hypertension in dogs produced by anastomosis of the systemic and pulmonary circulations46 have suggested that fibrinoid necrosis plays a critical role in the pathogenesis of plexiform lesions. Histologic and ultrastructural studies have demonstrated fibrin within the cytoplasm of the cells lining the vascular spaces.45 On the basis of these studies, it has been suggested that a focus of fibrinoid necrosis develops in a pulmonary artery branch adjacent to its site of origin. The high intravascular pressure causes this weakened vessel to dilate to form a sac or microaneurysm. Thrombus forms in the sac. This stimulates myofibroblasts to accumulate within the sac and secrete an extensive matrix of ground substance. This is the way that the typical plexiform arrangement of vascular channels and stroma is built up. The cells lining the vascular channels express factor VIII47 but differ from normal endothelium in that they lack a basement membrane, micropinocytotic vesicles, caveolae, and Weibel-Palade bodies. Instead their cytoplasm often contains numerous microfilaments of uniform thickness arranged in a haphazard pattern.45
Plexiform lesions tend to develop in arteries in which medial hypertrophy is not very prominent.27 It is possible that in the absence of maximal development of medial hypertrophy, the smooth muscle cells fail to accommodate a high transmural pressure and are more prone to fibrinoid necrosis. In this way, advanced medial hypertrophy may possibly give some protection against the formation of plexiform lesions.
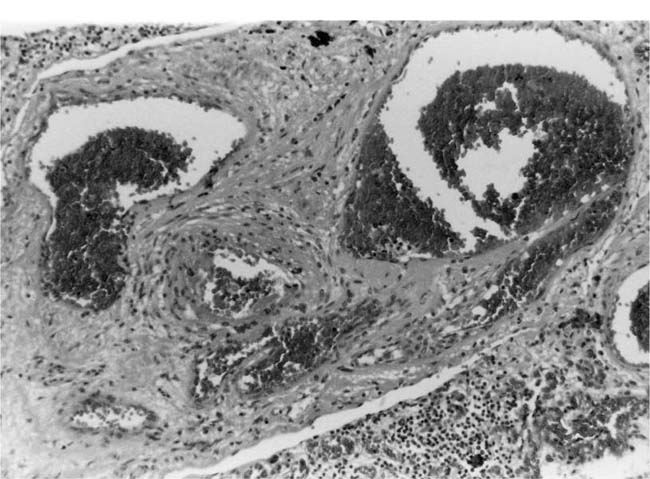
FIGURE 25–10 Muscular pulmonary artery showing an advanced plexiform lesion that contains large quantities of collagen deposited between the vascular channels. From the patient illustrated in Fig. 25–7 (H&E, ×200).
Dilatation Lesions
In severe hypertensive pulmonary vascular disease there may be pronounced dilatation of pulmonary arteries, often over a considerable length, leading to thinning of the arterial wall (Fig. 25–12). This may decrease the average thickness of the arterial media considerably. Such a dilated artery can constitute a diagnostic pitfall, because it may be misinterpreted as being normal and, hence, inconsistent with hypertensive pulmonary vascular disease. Extreme thinning of the walls of lateral branches of pulmonary arteries proximal to segments of obliterative concentric laminar intimal fibrosis may lead to the formation of dilatation lesions.48 Veinlike branches of muscular pulmonary arteries represent the extreme of vascular dilatation in which the media is all but absent (Fig. 25–13). The smooth muscle cells are attenuated and the elastic laminae are fragmented. The external elastic lamina is absent for most of its circumference so that the vessel resembles a pulmonary vein. These vein-like branches frequently form a convoluted cluster around the periphery of an obstructed pulmonary artery. Occasionally they form complex, large, compact structures that are termed angiomatoid lesions (Fig. 25–14). These dilatation lesions have been misinterpreted as arteriovenous anastomoses, but serial sections have demonstrated conclusively that they are arterial in nature and terminate in alveolar capillaries.40
Fibrinoid Necrosis and Arteritis
These uncommon lesions appear to be caused by a sudden and severe elevation of the intravascular pressure. Fibrinoid necrosis usually affects small muscular pulmonary arteries and pulmonary arterioles. It is commonly patchy in distribution with some vessels being severely affected whereas others are completely spared. Usually, the whole circumference of the artery is affected, but sometimes only a part of it is involved. Fibrinoid necrosis usually affects short segments of arteries and commonly occurs shortly after their origin from a parent artery. In the affected arteries, the media is swollen with loss of nuclear detail. It stains intensely and homogeneously with eosin and by the picro-Mallory and Martius scarlet blue methods for fibrin. The lumen frequently contains a thrombus. Fibrinoid necrosis may be the sole lesion in a hypertensive pulmonary artery, but it is frequently complicated by an infiltrate of neutrophil polymorphs in the media. In rare cases of pulmonary hypertension, arteritis may occur in the absence of fibrinoid necrosis. There is degeneration and some loss of smooth muscle cells but the striking change is an infiltrate of neutrophil polymorphs, which affects all layers of the wall and usually extends beyond the adventitia into the surrounding lung tissue (Fig. 25–15). Older lesions undergo organization with the development of granulation tissue, which involves the adventitia as well as the media. There is often fragmentation of the elastic laminae (Fig. 25–16).
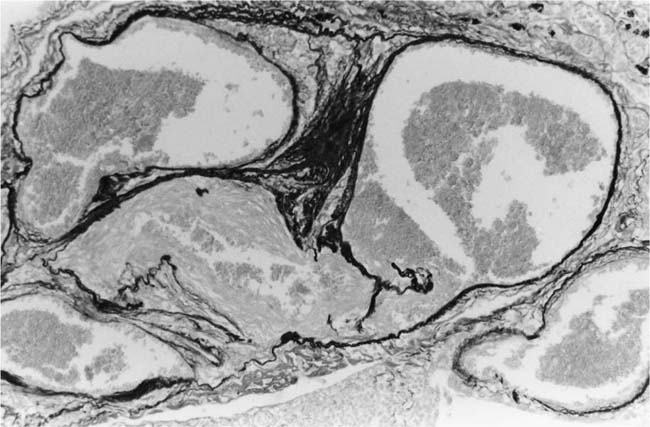
FIGURE 25–11 Muscular pulmonary artery showing a mature plexiform lesion. The vessel consists of a dilated sac containing irregular bands of fibroelastic tissue in which a plexus of vascular channels persists. From the same vessel illustrated in Fig. 25–10 (EVG, ×200).
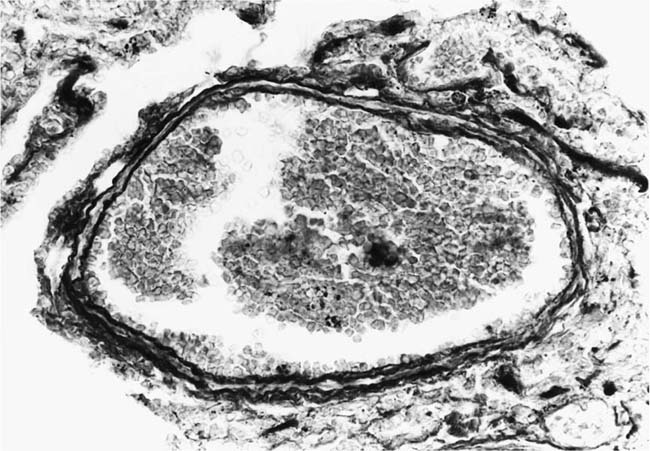
FIGURE 25–12 Muscular pulmonary artery from the same patient as in Fig. 25–6. Dilatation of the vessel has caused thinning of the media so that it resembles a normal pulmonary artery (EVG, ×500).
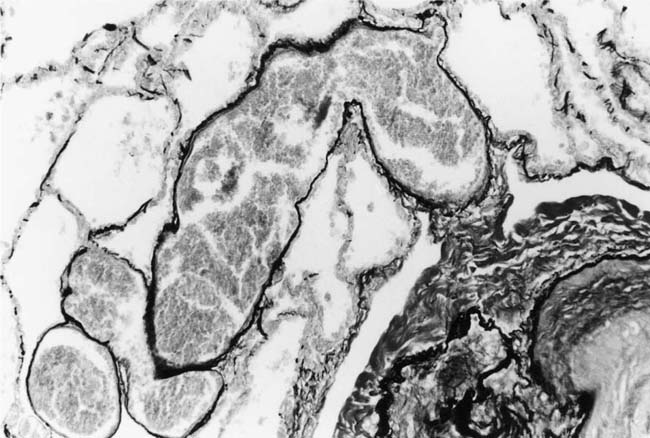
FIGURE 25–13 In the center of the picture is a veinlike branch of the muscular pulmonary artery shown in the bottom right corner. From the patient illustrated in Fig. 25–7 (EVG, ×200).
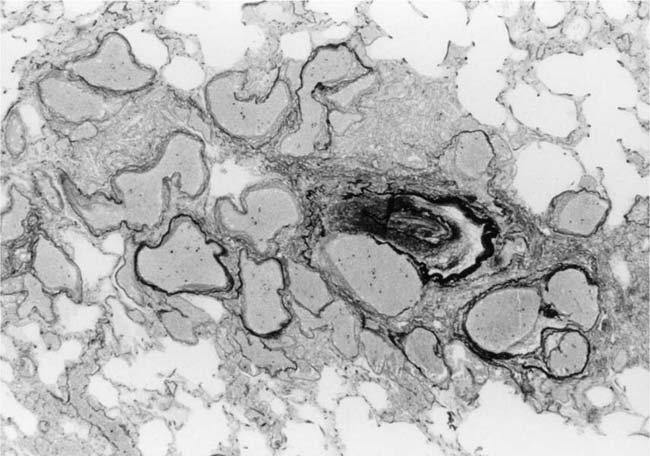
FIGURE 25–14 Just right of center is a muscular pulmonary artery that is occluded by intimal fibroelastosis. It is surrounded by large dilated vascular channels with thin walls. This is an example of an angiomatoid lesion. From the patient illustrated in Fig. 25–7 (EVG, ×80).
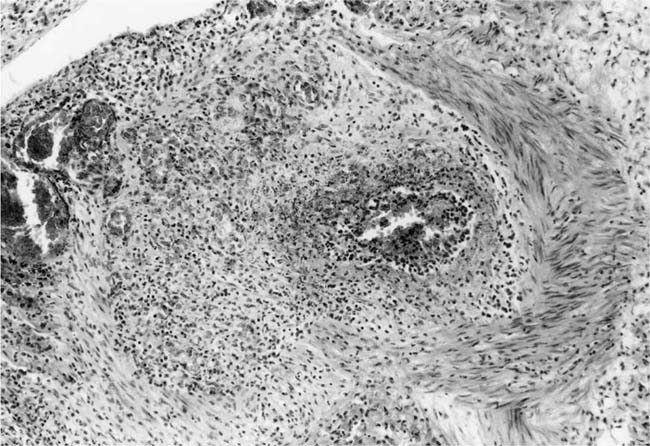
FIGURE 25–15 Muscular pulmonary artery from the patient illustrated in Fig. 25–6. It shows acute arteritis. In the right half of the figure, the media is intact. In the left half it is necrotic and infiltrated by neutrophil polymorphs that have extended into the adventitia (H&E, ×200).
In pulmonary hypertension, the pathogenesis of fibrinoid necrosis appears to be the same as in systemic hypertension.49 The high intravascular pressure creates small splits in the endothelium through which blood plasma passes into the subendothelial space, often distending it. The fibrin then becomes precipitated as lens-shaped crystalline structures. It is usually held up by the internal elastic lamina but may pass through fenestrae into the media, which it traverses until it is once again held up by the external elastic lamina. Because fibrinoid necrosis is dependent on the passage of fibrinogen into the media and not on the death of smooth muscle cells, the term fibrinoid necrosis is a misnomer.
Other Nonspecific Changes
A nonspecific feature of hypertensive pulmonary vascular disease is tortuosity of arterial branches. In histologic sections, coiled, multiple cross sections of arteries may lie in rows or clusters.
Generalized dilatation of muscular pulmonary arteries occurs in the presence of a large pulmonary blood flow, particularly in patients with an atrial septal defect. This may be difficult to assess histologically when comparing the caliber of an artery with the lumen of the associated bronchus or bronchiole. However, when all arteries available for examination are wider than the adjacent bronchi or bronchioles, generalized dilatation is probably present. Morphometry in such cases has revealed that although the amount of medial smooth muscle is increased, the average medial thickness of muscular pulmonary arteries expressed as a percentage of the external diameter is often within normal limits, implying that medial hypertrophy has been masked by the dilatation.50
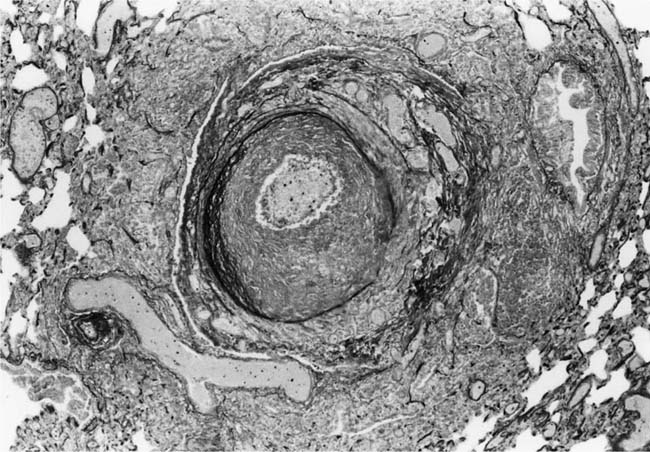
FIGURE 25–16 Muscular pulmonary artery from the patient illustrated in Fig. 25–6. It shows arteritis with organization. There is disruption of the internal and external elastic laminae with the development of granulation tissue involving the adventitia as well as the media (EVG, ×80).
Longitudinal smooth muscle cells are commonly encountered in muscular pulmonary arteries in hypertensive pulmonary vascular disease of any severity.51 This is a nonspecific change that is not related to the underlying cause of the pulmonary hypertension. However, it is frequently seen in patients with congenital heart disease over the age of 11 years.52 The smooth muscle cells are arranged in bundles or layers, particularly in the intima where they are situated within a reduplication of the internal elastic lamina (Fig. 25–17). Longitudinal smooth muscle cells may also be encountered within the media or adventitia.
Fresh and organizing thrombi are encountered in the muscular pulmonary arteries of ~30% of cases of congenital heart disease.52 Presumably, they are secondary to endothelial injury caused by the hemodynamic disturbances.53
Pulmonary Veins
In almost all patients over the age of 5 years with congenital heart disease, the pulmonary veins show either a fibrous intimal proliferation or fibrocellular intimal proliferation.52,54 Many patients show muscularization of the pulmonary veins with the development of a medial coat of smooth muscle separated by two elastic laminae. This latter change has been identified in children as young as 2 years of age. It is possible that these venous lesions are the consequence of increased levels of pulmonary blood flow, which may be especially high in an atrial septal defect.48
Lung Parenchyma
Lung parenchymal lesions, which have been described in patients with pulmonary hypertension due to systemic-to-pulmonary shunts, include increased numbers of alveolar macrophages,52 intraalveolar hemorrhage,52 hemosiderin-laden macrophages,48,52 focal intraalveolar fibrosis,52 cholesterol granulomas,52 mast cell hyperplasia,55 increased endocrine cells in the distal bronchioles,56,57 peribronchial lymphoid aggregates,52 and hypertrophy of bronchiolar smooth muscle (Table 25–5).57
The literature contains contradictory data about the occurrence of pulmonary hemosiderosis in patients with pulmonary hypertension due to systemic-to-pulmonary shunts. Pulmonary hemosiderosis was noted to be absent in a detailed pathologic and hemodynamic study of 18 patients with severe pulmonary hypertension due to congenital heart disease.58 One of the authors of this paper collaborated in a later study that revealed pulmonary hemosiderosis in 16 of 20 patients with severe pulmonary hypertension due to congenital heart disease.52 The difference in results may be attributable to a difference in the source of the tissue studied: the former paper was based upon tissue obtained at biopsy and autopsy, whereas the latter paper was based on tissue derived from heart–lung transplantation. In my experience, hemosiderosis is not usually encountered in the lungs of patients with pulmonary hypertension due to systemic-to-pulmonary shunts.
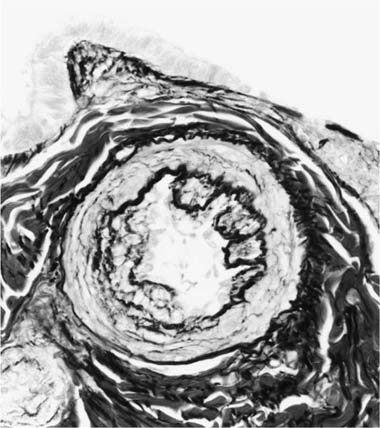
FIGURE 25–17 Muscular pulmonary artery from the 10-month-old infant illustrated in Fig. 25–4. There is severe medial hypertrophy. The intima includes large bundles of longitudinal smooth muscle cells situated within a reduplication of the internal elastic lamina (EVG, ×500).
Potentially reversible lesions |
Medial hypertrophy |
Cellular intimal proliferation |
Mild concentric laminar intimal fibrosis |
Intimal longitudinal muscle |
Irreversible lesions |
Severe concentric laminar intimal fibrosis |
Plexiform lesions |
Dilatation lesions |
Fibrinoid necrosis |
Electron microscopy has shown fibrosis of the alveolar walls without thickening of the capillary basement membrane in patients with congenital heart disease.59 The foci of intraalveolar fibrosis and cholesterol granulomas encountered in patients with pulmonary hypertension treated by heart–lung transplantation are thought to represent the end result of bleeding into the lungs.52 The hyperplasia of mast cells is also believed to be related to organizing hemorrhage and fibrosis.55 Cholesterol granulomas were originally considered to be specifically related to pulmonary hypertension, but they are now regarded as nonspecific lesions that may be found in a wide variety of lung diseases.60,61
The number of neuroendocrine cells has been noted to be increased in the distal bronchioles of patients with pulmonary hypertension due to congenital heart disease.56 In a group of patients who developed postoperative pulmonary hypertension following surgery for congenital heart disease, an increase in neuroendocrine cells was associated with hypertrophy of bronchiolar smooth muscle.57 Pulmonary neuroendocrine cells contain a variety of amines and peptides. It has been suggested that these substances may mediate constriction and hypertrophy simultaneously in bronchiolar and vascular smooth muscle.57
Relation Between Structural Changes, Hemodynamics, and Prognosis
In patients with post-tricuspid shunts such as a ventricular septal defect, the level of pulmonary artery pressure rises with the progressive severity of the hypertensive pulmonary vascular disease. Thus, the mean pulmonary artery pressure ranges from 40 to 70 mm Hg in patients with medial hypertrophy of muscular pulmonary arteries, and from 50 to 80 mm Hg in those with medial hypertrophy and intimal fibrosis.62–64 Mean pulmonary artery pressures ranging from 80 to 100 mm Hg are encountered in patients with plexiform lesions, dilatation lesions, and fibrinoid necrosis.62
In patients with medial hypertrophy with or without intimal fibrosis, the pulmonary blood flow is abnormally high. In patients with plexiform lesions, the pulmonary blood flow is either normal or diminished. The pulmonary blood flow is lower than normal in patients with dilatation lesions and fibrinoid necrosis.62
The pulmonary vascular resistance is only slightly elevated in patients with medial hypertrophy; when it is complicated by intimal fibrosis, there is a moderate increase in pulmonary vascular resistance to 3 times normal. In patients with dilatation lesions, the vascular resistance has increased by as much as seven to nine times. The highest pulmonary artery pressures and resistances are encountered in patients with fibrinoid necrosis where the resistance may be 15 times that of normal.62
Closure of Systemic-to-Pulmonary Shunts
Advances in surgery have enabled congenital cardiac defects to be repaired with minimal immediate mortality. However, the long-term prognosis following such operations is not necessarily good. For example, patients with a ventricular septal defect may have such a high pulmonary vascular resistance that they develop a reversal of the shunt. In this situation, the defect is acting as a safety valve, permitting blood entering the right ventricle to bypass the pulmonary vasculature. Closure of the defect diverts all the blood through the lungs, thereby increasing the pulmonary artery pressure rather than lowering it. One can envisage an exacerbation of pulmonary hypertension in patients with irreversible lesions in the pulmonary arteries. There have been studies to determine preoperatively the reversibility of pulmonary hypertension using pulmonary vasodilatory maneuvers such as the breathing of oxygen62,63 and the administration of acetylcholine or tolazoline.10 Such efforts are often successful, but sometimes they fail because these measures do not remove the main stimulus for vasoconstriction, which is high pressure, whereas surgery does reduce the pressure. The relation between arterial lesions in open lung biopsy specimens and the reversibility of pulmonary hypertension has been examined in patients studied immediately after the closure of ventricular and atrial septal defects,63 and 24 hours and 1 year after operation.54,64 Such studies have shown that medial hypertrophy and intimal fibrosis of muscular pulmonary arteries are associated with pulmonary hypertension of which is potentially reversible, whereas the presence of plexiform lesions and dilatation lesions is associated with high fixed levels of pulmonary vascular resistance and irreversible pulmonary hypertension.
The presence of potentially reversible pulmonary vascular disease is not always synonymous with operability and immediate postoperative survival.65,66 Infants and young children with congenital heart disease and severe medial hypertrophy of the muscular pulmonary arteries are prone to develop pulmonary hypertensive crises, which may cause death during surgery or in the postoperative period.54,65,66 These crises characteristically occur immediately after, or during the first few days following intracardiac repair in young children with a high preoperative pulmonary blood flow. Following surgery for congenital heart disease, there is often an increased reactivity of the pulmonary vasculature, resulting in sudden increases in pulmonary vascular resistance and pulmonary artery pressure. Pulmonary hypertensive crises are precipitated by hypoxia, but frequently occur in well-oxygenated patients for reasons that are unclear. In some patients, the episodes of postoperative pulmonary hypertension are accompanied by increases in respiratory system resistance of sufficient magnitude to interfere with mechanical ventilation. Such patients have hypertrophy of bronchiolar smooth muscle in addition to severe medial hypertrophy of muscular pulmonary arteries.57
Reversibility of Pulmonary Arterial Lesions
It has been possible to study the regression of hypertensive pulmonary arterial lesions in patients with congenital heart disease in whom banding of the pulmonary artery was performed (Table 25–5). Two lung biopsy specimens were available in these patients, one taken at the time of banding and the other several years later at the time of corrective surgery.67,68 From such investigations it appears that intimal longitudinal muscle and medial hypertrophy are largely reversible. Sometimes medial hypertrophy regresses completely, but often residual medial thickening is present several years after the banding procedure. In patients with an extremely thick media, where the mean medial thickness exceeds the internal diameter of the vessel, banding of the pulmonary artery may not lead to regression of the medial hypertrophy.69,70 Cellular intimal proliferation, even when it is severe, tends to regress, leaving only a thin residual layer of intimal fibrosis. Mild concentric laminar intimal fibrosis, occluding less than 20% of the average arterial lumen, is reversible.68 Severe concentric laminar intimal fibrosis is irreversible and even shows a tendency to progression. Plexiform lesions, dilatation lesions, and fibrinoid necrosis are irreversible.
Lung Biopsy in Systemic-to-Pulmonary Shunts
In most patients with congenital heart disease, measurements of the pulmonary vascular resistance and calculation of the ratio between the pulmonary vascular resistance and the systemic vascular resistance give reliable indications of the state of the pulmonary vascular bed, obviating the need for routine surgical lung biopsies.66 If a reliable assessment of the pulmonary vascular resistance cannot be made, it may be necessary to perform a lung biopsy to evaluate the state of the pulmonary arteries and decide whether or not the defect should be repaired.7,66,70,71 A biopsy specimen measuring 2.5 × 2 × 1 cm from an adult lung, or a proportionately smaller specimen from a child, will usually yield tissue containing pulmonary arteries that are representative of the lungs as a whole, providing the depth is not much less than 2 cm.3,7 The lingula of the left upper lobe is a favorite site for biopsy because of its accessibility. It has been suggested that the lingula is an inappropriate site for biopsy because the pulmonary arteries in this segment have thicker medial and intimal layers.32,72 However, other studies have failed to observe any significant differences between the pulmonary arteries in various parts of the lungs, including the lingula.73,74 Accordingly, there seems to be no good reason why the lingula should be excluded as a biopsy site in patients with congenital heart disease. A surgical lung biopsy may yield valuable information, but it should not be undertaken without careful consideration of the risks involved. Mortality and morbidity rates of 20% and 13%, respectively, have been reported when an open lung biopsy has been used as an isolated procedure in children with congenital heart disease.66 Intraoperative frozen sections are inadequate to assess hypertensive pulmonary vascular disease because of the necessity for multiple well-stained sections.3,7,66
Grading of Hypertensive Pulmonary Disease
In 1958 Heath and Edwards48 introduced a system for grading hypertensive pulmonary vascular lesions in patients with congenital heart disease, based on progressive changes in the muscular pulmonary arteries and pulmonary arterioles (Table 25–6). The lung tissue sample is assigned a grade that corresponds with the most severe lesion present. The grade of hypertensive pulmonary vascular disease was found to correlate with the severity of pulmonary hypertension62 and the immediate reversibility of pulmonary hypertension following closure of ventricular and atrial septal defects.63 In a follow-up of patients who had closure of a ventricular septal defect performed over 25 years previously, the Heath-Edwards grade was found to correlate well with the long-term clinical or hemodynamic status of the patient in 93% of cases.75 Grade 4 changes usually predicted a fatal outcome, whereas grade 1 and grade 2 changes generally did not.
Grade 1: | Medial hypertrophy of muscular pulmonary arteries; muscularization of pulmonary arterioles; no intimal changes |
Grade 2: | Medial hypertrophy; cellular intimal proliferation |
Grade 3: | Medial hypertrophy; intimal fibrosis |
Grade 4: | Occlusive intimal fibrosis; generalized dilatation; plexiform lesions |
Grade 5: | Vein-like branches; angiomatoid lesions |
Grade 6: | Fibrinoid necrosis; arteritis |
The Heath-Edwards grading system has been criticized on the grounds that fibrinoid necrosis of the pulmonary arteries may precede development of plexiform lesions, but this occurrence does not invalidate the principle of the grading system because the presence of fibrinoid necrosis, dilatation lesions, and plexiform lesions all signify an advanced form of hypertensive pulmonary vascular disease associated with irreversible pulmonary hypertension. No one has been able to show that any one of the three advanced grades indicates a prognosis different from that of the other two. A problem with the Heath-Edwards grading system that emerged over 25 years after it was introduced is that it does not distinguish between mild and severe intimal fibrosis.7,68 This distinction is of practical importance because mild concentric laminar intimal fibrosis has been found to be a reversible lesion, whereas severe concentric laminar intimal fibrosis is irreversible and even shows a tendency to progression.68
Other grading systems based on various combinations and permutations of medial thickening, intimal thickening, and plexiform lesions have been proposed. However, unlike the Heath-Edwards grading system, they have not been validated by a long-term follow-up. A grading system has been proposed that assesses (1) extension of muscle into smaller and more peripheral arteries than normal, (2) abnormal extension of muscle plus an increase in medial thickness, and (3) grade B changes plus a reduction in the number of small pulmonary arteries.2 The validity of this grading system has been questioned because of the inability to confirm a reduction in the number of small pulmonary arteries in pulmonary hypertension.4,7,8
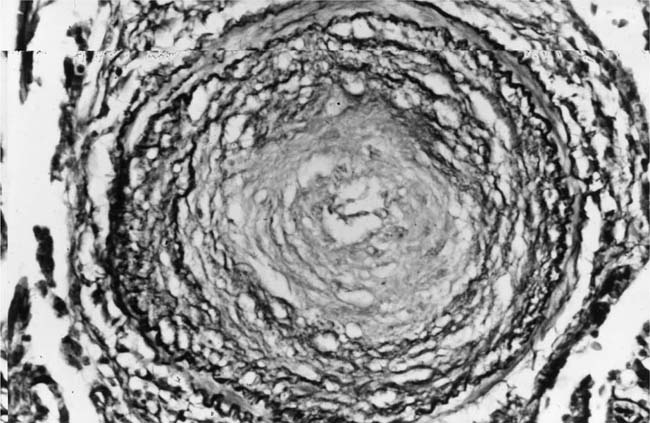
FIGURE 25–18 Muscular pulmonary artery showing severe narrowing of the lumen due to concentric laminar intimal fibrosis. From a patient with primary pulmonary arterial hypertension (EVG, ×500).
Primary Pulmonary Arterial Hypertension
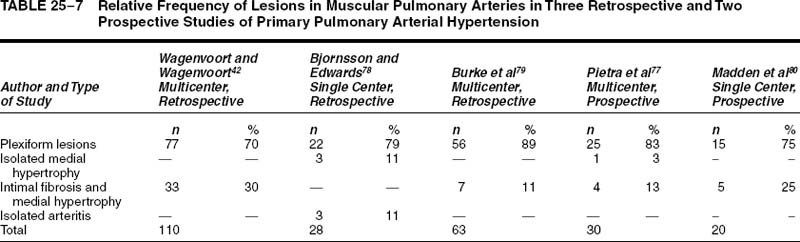
The term primary pulmonary arterial hypertension is used here to denote patients with pulmonary hypertension of unknown cause, in whom the significant vascular lesions are confined to the pulmonary arteries. Thus, patients with pulmonary veno-occlusive disease and chronic thrombotic or embolic disease are excluded from this rubric. Primary pulmonary arterial hypertension, initially described by Dresdale et al76 in 1951, most commonly occurs in young adult females. In one prospective77 and two retrospective78,79 studies, the mean age ranged from 30 to 32 years. However, the condition has been identified in infants and adults in their 7th decade. In the three series, the male-to-female ratio ranged from 1:1.3 to 1:3.2. This female predominance was evident both in adults and children.
The pulmonary vascular and parenchymal lesions that occur in patients with primary pulmonary arterial hypertension are similar to those that are found in subjects with systemic-to-pulmonary shunts.52 The muscular pulmonary arteries may show medial hypertrophy, concentric laminar intimal fibrosis (Fig. 25–18), plexiform lesions (Figs. 25–19 and 25–20), dilatation lesions, fibrinoid necrosis, and arteritis. In 1975, an expert committee of the WHO recommended that this pattern of hypertensive pulmonary vascular disease be designated plexogenic pulmonary arteriopathy.12 Plexogenic pulmonary arteriopathy was envisaged as a disease with a sequence of changes that may eventually include plexiform lesions. Plexogenic pulmonary arteriopathy is a subtle term that implies the potential to develop plexiform lesions. According to the WHO, it is permissible to diagnose plexogenic pulmonary arteriopathy in the absence of plexiform lesions. Using this criterion, only 70 to 89% of the cases designated plexogenic pulmonary arteriopathy actually have plexiform lesions.42,77–80 The remaining cases have either medial hypertrophy and intimal fibrosis, or isolated medial hypertrophy (Table 25–7). The recommendation of the WHO has been either misunderstood or misinterpreted by many pathologists and clinicians, resulting in much confusion in the literature. Some authors have restricted the diagnosis of plexogenic pulmonary arteriopathy to cases in which plexiform lesions are actually present.78 To add to the confusion, reference has even been made to preplexiform lesions.80 It is desirable for pathologists to make morphologic diagnoses based on what they see, rather than speculating on what might develop later. Accordingly, the consultant pathologists associated with the National Heart, Lung, and Blood Institute Registry of Primary Pulmonary Hypertension recommended the use of the term pulmonary arteriopathy with plexiform lesions.77 The major criterion for making this diagnosis is the identification of plexiform lesions in histologic sections.
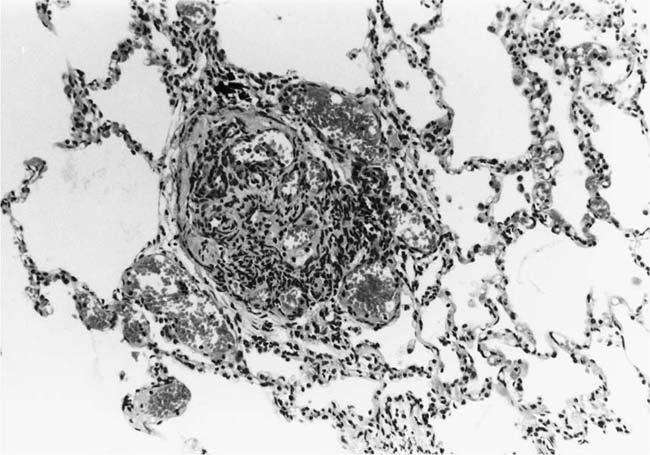
FIGURE 25–19 Muscular pulmonary artery showing a plexiform lesion. From a 42-year-old woman with primary pulmonary arterial hypertension (H&E, ×200).
Muscular Pulmonary Arteries and Pulmonary Arterioles
The frequency with which plexiform lesions, isolated medial hypertrophy, and medial hypertrophy with intimal fibrosis were found in three retrospective and two prospective studies is shown in Table 25–7. The pattern of pulmonary vascular disease that was most commonly encountered was pulmonary arteriopathy with plexiform lesions. Its incidence in the three retrospective studies ranged from 70 to 89%, whereas in the two prospective studies it ranged from 75 to 84%. The incidence of isolated medial hypertrophy ranged from 0 to 11%, whereas the incidence of medial hypertrophy and intimal fibrosis ranged from 0 to 30%. One retrospective series was unusual in that it included three cases (11%) of isolated pulmonary arteritis.78 These were characterized by a severe nongranulomatous pulmonary arteritis with obstructive secondary thrombosis, but without plexiform lesions, pulmonary phlebitis, systemic vasculitis, or collagen vascular disease.
Perhaps the most accurate estimate of the relative frequency of arterial lesions may be obtained from the National Heart, Lung, and Blood Institute Registry of Primary Pulmonary Hypertension. The purpose of the registry was to analyze in a systematic and prospective manner the natural history, epidemiology, pathology, and treatment of unexplained pulmonary hypertension in a cohort of patients in whom the diagnosis was established by standardized clinical and laboratory criteria.11,81 Twenty-five of 30 patients with primary pulmonary arterial hypertension had pulmonary arteriopathy with plexiform lesions (83%), 4 patients had medial hypertrophy and intimal fibrosis (13%), and 1 patient had isolated medial hypertrophy (3%).77 It has been suggested that cases of isolated medial hypertrophy, and intimal fibrosis and medial hypertrophy, represent early forms of primary pulmonary arterial hypertension in which plexiform lesions have not yet developed.82 This is a potentially important group because it may represent patients who are amenable to drug therapy due to the absence of severe obstructive and irreversible lesions.78
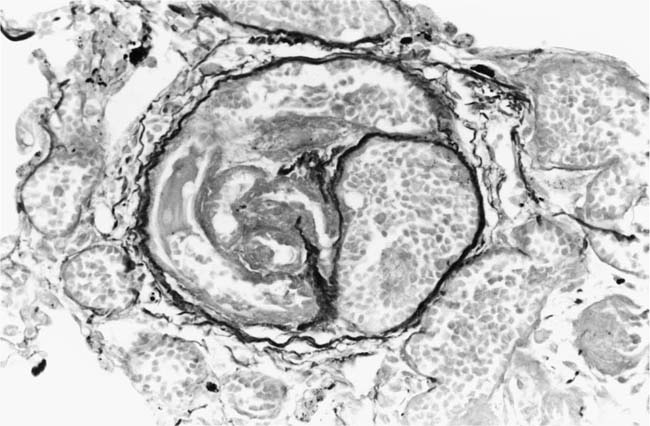
FIGURE 25–20 Mature plexiform lesion in a transverse section of a muscular pulmonary artery from the patient illustrated in Fig. 25–19. In this dilated segment, there is destruction of the arterial wall with disappearance of the internal elastic lamina. The sac contains irregular thin bands of fibroelastic tissue in which a plexus of vascular channels persists (EVG, ×500).
There is ultrastructural evidence that the cellular intimal proliferation is derived from smooth muscle cells from the inner half of the tunica media that migrate into the intima through gaps in the internal elastic lamina.36,83 These gaps may be natural, or may be related to increased activity of endogenous vascular elastase.38,84 Immunohistochemical studies have suggested that smooth muscle cells in the media and neointima actively synthesize collagen in patients with severe primary pulmonary arterial hypertension, and that transforming growth factor-β may modulate this process.85 Fibrosis also occurs in the adventitia of the muscular pulmonary arteries.86 Muscularization of pulmonary arterioles occurs but is less common than in patients with systemic-to-pulmonary shunts.52 Plexiform lesions are relatively uncommon; they involve 3.9% of arteries in biopsy tissues, and 7% of arteries in autopsy tissues.77 In contrast to systemic-to-pulmonary shunts, the majority are associated with intraacinar arteries.39 In 70% of cases, the plexiform lesions are scantily infiltrated by T cells, B cells, and macrophages.87 The endothelial cell proliferation in the plexiform lesions of primary pulmonary arterial hypertension is reported to be monoclonal, in contrast to secondary pulmonary hypertension where it is polyclonal.47,88 There is no correlation between the numbers of plexiform lesions and the pulmonary artery pressure.43,78,79 In two retrospective studies, dilatation lesions were identified in 50% and 73% of patients, respectively.78,79 However, in a prospective study, dilatation lesions were rarely seen.77 Fibrinoid necrosis and arteritis are uncommon lesions occurring in ~11% of patients.79
Thrombotic lesions have been reported as occurring in the pulmonary arteries in 27% to 65% of patients with primary pulmonary arterial hypertension.52,77,79,80 They are absent or scarce in children, but common in adults. The number of organized and recanalized thrombi in tissue sections appears to be related to the duration of the illness, but not the stage of the disease. The occurrence of thrombi is independent of the presence of plexiform lesions.89 Thrombotic lesions occur with equal frequency in pulmonary vascular disease secondary to systemic-to-pulmonary shunts.52 Apparently, the combination of sustained pulmonary hypertension and age, possibly through endothelial injury, may elicit thrombosis and its sequelae, which in turn may aggravate the pulmonary arterial pressure.
Longitudinal smooth muscle cells are commonly seen in muscular pulmonary arteries in patients with primary pulmonary arterial hypertension.52 The smooth muscle cells are arranged in bundles or layers, particularly in the intima where they are situated within a reduplication of the internal elastic lamina. Longitudinal smooth muscle cells may also be seen in the media or adventitia.
Pulmonary Veins
In the majority of patients with primary pulmonary arterial hypertension, the pulmonary veins show a nonocclusive fibrocellular or fibrous intimal proliferation, and adventitial fibrosis.52,78,79,86 In a few cases, this is accompanied by the development of a thick medial coat of smooth muscle separated by two elastic laminae.52
Lung Parenchyma
The lung parenchymal lesions that occur in primary pulmonary arterial hypertension are similar to those occurring in systemic-to-pulmonary shunts (Table 25–4), with the exception of hypertrophy of bronchiolar smooth muscle, which has not been described in this condition.52 Peripheral airways obstruction was described in 10 nonsmokers with primary pulmonary arterial hypertension. Autopsy and lung biopsy specimens from three of the cases showed narrowed small airways with thickened walls, infiltrated by lymphocytes, some plasma cells, and a few neutrophil polymorphs.90 In my experience, hemosiderosis is not usually encountered in the lungs of patients with primary pulmonary arterial hypertension. However, two studies in the literature quote incidences of 94%52 and 71%.79 In the latter study, 88% of the cases were autopsies and the authors do not mention whether or not they used the Perls Prussian blue method to detect hemosiderosis. Pigment-laden macrophages are commonly encountered within the lungs, and it is easy to overdiagnose hemosiderosis if a special stain to detect ferric iron is not performed. The former study was based on tissue derived from heart–lung transplantation; the authors did stain sections by the Perls Prussian blue method.
Hemodynamics and Prognosis
In one of the retrospective studies involving 19 patients with primary pulmonary arterial hypertension, the pulmonary artery pressure ranged from 47 to 99 mm Hg with a mean (± standard deviation, SD) of 73 ± 14 mm Hg.78 The mean survival of 22 patients was 63 months, and 45% died suddenly. In another retrospective study involving 41 patients with primary pulmonary arterial hypertension, the mean pulmonary artery pressure was 66 mm Hg, and the mean survival time was 36 months.79 In the Registry of Primary Pulmonary Hypertension, the mean pulmonary artery pressure (± SD) in the 20 patients with plexiform lesions was 76.4 ± 22.2 mm Hg.77 They had a high pulmonary vascular resistance (44.5 ± 22.4 units), and the low cardiac output (1.7 ± 0.41 L/min/m2). The mean and median survival times were 297 days and 102 days, respectively. Although the prognosis for most patients with primary pulmonary arterial hypertension is poor, occasional cases of regression91 and spontaneous resolution have been reported.92
Etiology and Pathogenesis
By definition, the cause of primary pulmonary arterial hypertension is unknown. Two dissimilar processes appear to be associated with its onset.52 One factor appears to be constriction of small pulmonary arteries leading to the development of medial hypertrophy. A second factor is the migration of smooth muscle cells from the media of muscular pulmonary arteries through defects in the internal elastic lamina into the intima, where they proliferate and occlude these vessels. These observations have therapeutic implications.52,78 Pulmonary vasodilators may influence the vasoconstrictive element, but control of the intimal proliferation may require therapies that influence the growth and differentiation of smooth muscle cells. Unfortunately, the intimal proliferation may be a very early feature of the disease, likely to have an adverse effect on therapeutic measures from the onset. Factors that need to be considered in the pathogenesis of primary pulmonary arterial hypertension include pulmonary vascular hyperreactivity, genetic factors, hormonal factors, autoimmune disease, and thrombosis.
Pulmonary Vascular Hyperreactivity
It has been suggested that some individuals have hyperreactive pulmonary blood vessels, and that patients with primary pulmonary arterial hypertension respond with vasoconstriction to various stimuli that would have little or no effect in normal people.3,42 There have been occasional reports that primary pulmonary arterial hypertension is more common at high altitude than at sea level. Exposure to hypoxia may initiate the development of pulmonary hypertension in subjects with hyperreactive pulmonary vascular beds.
Genetic Factors
The literature includes numerous examples of the familial occurrence of this condition, usually concerning parent–child relationships, or siblings who are sometimes twins.42,93 Six percent of the patients entered into the National Heart, Lung, and Blood Institute Registry of Primary Pulmonary Hypertension were examples of familial primary pulmonary arterial hypertension (disease affecting a first-order blood relative).11 Patients who had positive family histories were usually diagnosed sooner after the onset of symptoms than were the other registry patients. There were no differences, however, in their ages, hemodynamic findings, or survival. Twenty-one families in the United Kingdom and nearly 100 families in the United States have now been identified in which the disease occurs in at least two first-degree relatives.94,95 Further kindreds have been described in Europe, Japan, Russia, and Australia. The gene for familial primary pulmonary arterial hypertension, currently known as PPH1, is transmitted as an autosomal trait. The overall penetrance of the gene (the chance of developing the disorder in individuals who carry the gene) is estimated to be about 10 to 20%; the penetrance of the gene is higher in females (30%) than in males (15%).95 The cause of this gender difference is unknown. Father-to-son transmission has been shown in multiple families. The incidence of familial primary pulmonary arterial hypertension in the United States is probably higher than the 6% of cases reported from the registry; most sporadic cases of primary pulmonary arterial hypertension probably represent familial disease.95 The low penetrance of the gene coupled with an incomplete family history may hide its inheritance. Generations can be skipped. Decreasing age of onset of disease in subsequent generations has been described. This worsening of familial disease in subsequent generations is known as genetic anticipation. Genetic linkage studies have identified a disease locus on chromosome 2q33.96 Mutations of the gene encoding bone morphogenic protein receptor 2(BMPR2) have been identified in ~50% of families with familial primary pulmonary arterial hypertension, and 26% of patients with sporadic primary pulmonary arterial hypertension94–96 Bone morphogenic proteins are members of the transforming growth factor-β (TGF-β) superfamily of signaling molecules. Originally discovered as potent inducers of bone and cartilage formation, bone morphogenic proteins regulate cell differentiation, proliferation, and apoptosis in most organs and tissues of the body. It is speculated that mutations in BMPR2 interrupt the bone morphogenic protein signaling pathway, resulting in proliferation, rather than apoptosis of cells within small pulmonary arteries. The failure to find BMPR2 mutations in all families with familial primary pulmonary arterial hypertension, and in all patients with sporadic primary pulmonary arterial hypertension, suggests that other genes or etiologic factors remain to be identified.96 The development of primary pulmonary arterial hypertension may require “two hits.” The first hit could be a mutation in BMPR2; the second hit could be mutation of another gene, or an environmental factor such as exposure to a drug (e.g., fenfluramine) or a virus (e.g., human immunodeficiency virus).97
Hormonal Factors
It has been suggested that hormonal factors may play a role in the etiology of primary pulmonary arterial hypertension because of the tendency of the disease to develop in young adult females. Neither oral contraceptive use nor pregnancy appear to be etiologic factors, because their frequencies in patients with primary pulmonary arterial hypertension are similar to those of the general population.11 In an international epidemiologic study, neither oral contraceptive nor estrogen use was associated with a significantly increased risk of pulmonary hypertension.98 Although not currently recognized as risk factors for primary pulmonary arterial hypertension, it has been suggested that oral contraceptives and hormone replacement therapy should be used with caution in patients and families with known primary pulmonary arterial hypertension, because the interactions of these hormones with BMPR2 are unknown.95,96 Pregnancy may exacerbate preexisting primary pulmonary arterial hypertension, but its development during pregnancy is rare.
Autoimmune Disease
Several studies of patients with primary pulmonary arterial hypertension have shown evidence of an associated autoimmune disease. Thus, Raynaud’s phenomenon and serum antinuclear antibodies were detected in 11% and 29%, respectively, of 187 patients in a prospective study of primary pulmonary arterial hypertension.81 Pulmonary arterial hypertension has been described in several autoimmune disorders such as systemic lupus erythematosus,99 scleroderma,100 rheumatoid disease,101 Sjögren’s syndrome,102 dermatomyositis, and polymyositis.103
Thrombosis
It has been suggested that in patients with primary pulmonary arterial hypertension, plexiform lesions and thrombotic lesions are not specific, but represent different manifestations of the same pathologic process.93 However, primary pulmonary arterial hypertension and chronic thrombotic and/or embolic pulmonary appear to be two distinct pathologic entities with different hemodynamic features and prognoses.52,77 Furthermore, plexiform lesions have never been described in autopsy studies of patients with chronic thrombotic or embolic disease.104,105 In a study purporting to demonstrate plexiform lesions in patients with chronic thromboembolic pulmonary hypertension,106 the illustrations depict recanalized thrombi rather than plexiform lesions.107 Further evidence for the existence of primary pulmonary arterial hypertension separate from chronic thrombotic or embolic disease is the demonstration of an increased number of pulmonary endocrine cells containing gastrin-releasing peptide in primary pulmonary arterial hypertension, but not in chronic thrombotic or embolic disease.52,56 Recanalized thrombi in the pulmonary arteries have been reported in up to 65% of patients with primary pulmonary arterial hypertension.52,77,79,80 The number per case is usually quite small.78 These thrombotic lesions are probably secondary phenomena related to hemodynamic factors89 because they occur with equal frequency in patients with congenital systemic-to-pulmonary shunts.52
Chronic Thrombotic or Embolic Pulmonary Hypertension
The terms thrombotic pulmonary arteriopathy3 and pulmonary arteriopathy with thrombotic lesions77 are preferable to the term thromboembolic pulmonary hypertension because it is impossible to distinguish between embolic thrombi and in situ thrombi by microscopy alone.78 Although the embolic nature of thrombotic pulmonary arteriopathy has been emphasized,42 sources of such emboli in the systemic veins and right side of the heart frequently cannot be demonstrated at autopsy.104,108 It has therefore been suggested that in some cases in situ thrombosis of the pulmonary arteries may occur.78,109,110
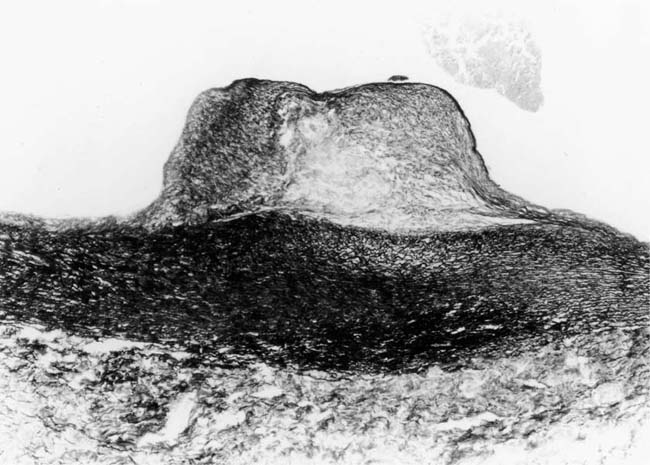
FIGURE 25–21 An intimal fibroelastic plaque protrudes into the lumen of an elastic pulmonary artery. It represents an organized mural thrombus (EVG, ×80).
Clinically apparent pulmonary embolism recurs in from 8 to 20% of patients treated for pulmonary thromboembolism.111 In view of these statistics, it is surprising that chronic pulmonary hypertension following pulmonary thromboembolism is such a rare condition with a prevalence at autopsy ranging from 0.15 to 0.38%.112 In one study, serial lung scans done in patients with acute pulmonary thromboemboli showed complete normalization of the perfusion defects in 86% of patients over a 1-year period, and it was estimated that less than 2% of these would develop chronic pulmonary hypertension.109 In a more recent study, 78 patients with acute pulmonary thromboembolism were subjected to a 1-year follow-up by serial echocardiography Doppler and a 5-year clinical follow-up. Four patients (5.1%) developed chronic pulmonary hypertension, and three underwent pulmonary thromboendarterectomy.113
Chronic pulmonary thrombotic or embolic pulmonary hypertension occurs in two forms108,109: occlusion of the proximal pulmonary arteries, and widespread occlusion of distal muscular pulmonary arteries. Some patients with occlusion of the proximal pulmonary arteries may complain of hemoptysis and recurrent episodes of pleuritic pain, but the majority present with increasing dyspnea on exertion.108,114 Patients with widespread thrombotic occlusion of the distal muscular pulmonary arteries may be clinically indistinguishable from patients with primary pulmonary arterial hypertension.
Thrombotic Obstruction of Proximal Pulmonary Arteries
Chronic thrombotic occlusion of the main pulmonary arteries is a rare entity with an incidence at autopsy ranging from 0.02 to 0.2%.104,112,115 In most instances, it is impossible at autopsy to distinguish with certainty between old organized thromboemboli and thrombi formed in situ. Some authors have denied the existence of primary thrombosis of the main pulmonary arteries and stated that the diagnosis is often made as a result of inadequate examination for possible sources of emboli.116 However, there are cases of thrombotic occlusion of the main pulmonary arteries in which no source for embolism could be discovered at autopsy.108
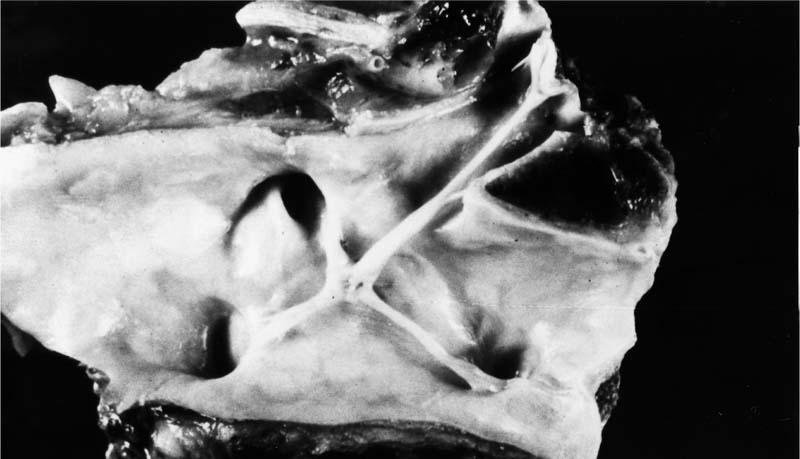
FIGURE 25–22 A pulmonary artery has been opened longitudinally to show fibrous bands stretching across the lumen. These represent an organized occluding thrombus.
At autopsy, the affected major pulmonary arteries are dilated and either stenosed or completely occluded by gray or yellow, firmly adherent laminated thrombus.104 On microscopic examination, small blood vessels and fibrous connective tissue attach the thrombus to the intimal surface and extend for a variable distance into the thrombus. The intima shows fibrous thickening, and the underlying media frequently shows basophilic degeneration resembling cystic medial necrosis.104 Non-occlusive thrombi tend to organize into fibrous intimal plaques (Fig. 25–21). If the vessel was occluded by the thrombus, the end result is likely to be a fibrous band (Fig. 25–22) stretching in a variety of somewhat bizarre patterns across the lumen of the artery.117 Such fibrous bands are relatively frequent in elastic pulmonary arteries (Fig. 25–23), and are a clear indication of previous thrombotic episodes. They may give rise to systolic murmurs over the lung fields,114 and can be visualized by pulmonary angiography and pulmonary angioscopy. In an autopsy study of 68 cases of thrombosis of the proximal pulmonary arteries, multiple organized thrombi were noted in the distal muscular pulmonary arteries in 65% of patients. In 35% of cases the distal muscular pulmonary arteries were microscopically normal; in 51% of cases they showed medial hypertrophy; in 13% of cases there was intimal proliferation.104 Plexiform lesions were not reported.
It is important to consider the diagnosis of thrombosis of the proximal pulmonary arteries in any patient who presents with dyspnea on exertion because this condition can be treated surgically.114 There is no plane of cleavage between the organized thrombus and the intima, so it is necessary to carry out thromboendarterectomy rather embolectomy. The surgical pathology specimen consists of partially organized pale red thrombus adherent to firm, resilient gray, organized thrombus that merges with the fibrotic intima (Fig. 25–24). The underlying media may include abnormal thick vascular channels representing bronchopulmonary arterial anastomoses (Fig. 25–25).
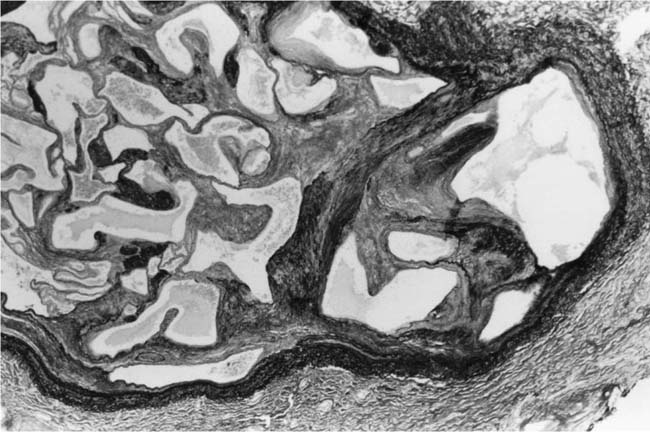
FIGURE 25–23 The lumen of this elastic pulmonary artery is occupied by a complex arrangement of bands of fibroelastic tissue representing an organized thrombus. From a 55-year-old man with chronic thromboembolic pulmonary hypertension. The free wall of his right ventricle weighed 115 g; the upper limit of normal is 80 g (EVG, ×50).
FIGURE 25–24 Surgical pathology specimen from pulmonary thromboendarterectomy. (Courtesy of Dr. F. V. Lobo.)
Hemodynamics and Prognosis
In 128 patients with thrombosis of the main pulmonary arteries, the mean pulmonary artery pressure was 47 ± 12 mm Hg. The mean pressure fell to 28 ± 9 mm Hg at 2 to 3 days after pulmonary thromboendarterectomy.114 In 47 patients who returned for follow-up 6 to 24 months after surgery, the mean pulmonary artery pressure was 24 ± 10 mm Hg. The operative mortality was 12%.
Thrombotic Obstruction of Distal Pulmonary Arteries
In two of the large retrospective studies of unexplained pulmonary hypertension, thrombotic obstruction of the distal pulmonary arteries was twice as common in women as in men. The mean ages of the patients in the two series were 44 and 41 years, respectively.42,78 In another large retrospective study of unexplained pulmonary hypertension, thrombotic obstruction of the distal pulmonary arteries was twice as common in men as in women and the mean age was 41 years.79 In a prospective study of unexplained pulmonary hypertension, there were nine men and 10 women with thrombotic obstruction of the distal pulmonary arteries.77 The mean age was 36 years.
In thrombotic obstruction of the distal pulmonary arteries, the muscular pulmonary arteries and pulmonary arterioles show cushionlike eccentric patches of intimal fibrosis (Fig. 25–26). When traced in serial sections, postthrombotic intimal fibrosis, although almost completely occlusive, often extends over relatively short distances (Fig. 25–27). Consequently, in random histologic sections, many pulmonary arteries show no intimal thickening at all, and occluded vessels are often uncommon (Fig. 25–28). When the amount of intimal fibrosis is assessed morphometrically, the average value is therefore usually low, compared with other forms of hypertensive pulmonary vascular disease in which intimal fibrosis affects long segments of the arteries.42 Recanalization may give rise to colander-like lesions (Fig. 25–29) and intravascular fibrous septa (Fig. 25–30). Fresh thrombus is usually considered to be rare,3,77 but in one of the retrospective studies of unexplained pulmonary hypertension, fresh thrombus was noted in 46% of cases of thrombotic obstruction of the distal pulmonary arteries.79
It has been stated that medial hypertrophy is either mild or absent,3 but it was observed in 64% of cases of thrombotic obstruction of the distal pulmonary arteries in one of the retrospective series of cases of unexplained pulmonary hypertension.78 Furthermore, in the prospective study of unexplained pulmonary hypertension, there was no significant difference in the frequency of medial hypertrophy when thrombotic pulmonary arteriopathy was compared with pulmonary arteriopathy with plexiform lesions.77 Morphometry of the media and intima failed to detect significant differences in arterial wall thickness between thrombotic pulmonary arteriopathy and pulmonary arteriopathy with plexiform lesions. Plexiform lesions and dilatation lesions have not been described in patients with thrombotic obstruction of the distal pulmonary arteries. Pulmonary arteritis rarely occurs.78 The pulmonary veins are normal. It is most unusual for a biopsy specimen to show evidence of infarction. Pulmonary hemosiderosis was identified in 10 of 13 cases of thrombotic obstruction of the distal pulmonary arteries in a retrospective study.79
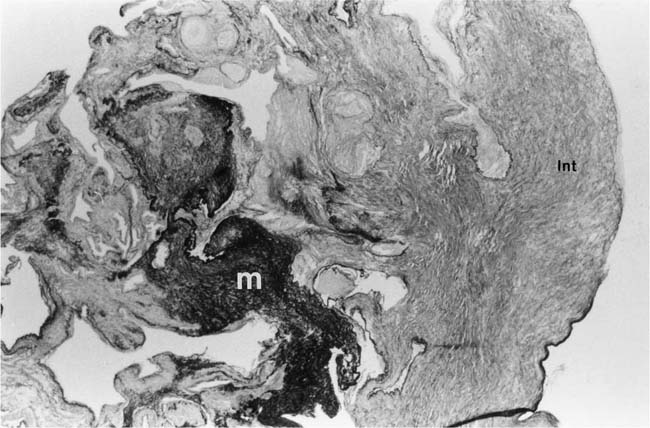
FIGURE 25–25 Surgical pathology specimen from pulmonary thromboendarterectomy in a 50-year-old man. The intima (Int) shows pronounced fibrous thickening. The elastic tissue of the media (m) is disrupted by numerous abnormal vascular channels, which are also present in the intima. These represent bronchopulmonary arterial anastomoses (EVG, ×50).
Hemodynamics and Prognosis
In one of the retrospective studies involving 40 patients with thrombotic obstruction of the distal pulmonary arteries, the pulmonary artery pressure ranged from 38 to 99 mm Hg with a mean (± SD) of 61 ± 12 mm Hg.78 The mean survival of 45 patients was 48 months with a range of 1 to 204 months. Eighteen percent of the patients died suddenly. In another retrospective study involving 13 patients, the mean pulmonary artery pressure was 48 mm Hg, and the mean survival 61 months.79 In a prospective study involving 10 patients with thrombotic obstruction of the distal pulmonary arteries, the mean pulmonary artery pressure was 60 ± 15 mm Hg.77 The mean and median survival times were 1070 days and 858 days, respectively.
Chronic Pulmonary Venous Hypertension
Any disease that is associated with chronic pulmonary venous hypertension may produce a characteristic combination of changes in the pulmonary arteries, alveolar capillaries, pulmonary veins, lymphatics, interstitial tissue, and alveoli (Table 25–8).118,119 This morphologic pattern has been called congestive vasculopathy.3
Chronic pulmonary venous hypertension may be caused by lesions of the pulmonary veins, left atrium, mitral valve, left ventricle, aortic valve, aorta, and mediastinum (Table 25–9). These lesions are usually acquired but may be congenital. Pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis are rare acquired disorders that cause obstruction of the intrapulmonary veins and venules. Obstruction of the large extrapulmonary veins is unusual: it may be congenital, as in anomalous pulmonary venous drainage and congenital stenosis of one or more major pulmonary veins or the venoatrial junction; it may also be acquired as a result of neoplasm or fibrosing mediastinitis causing extrinsic compression.120 Severe pulmonary vein stenosis has been described following catheter ablation of atrial fibrillation, but this has not yet been associated with pulmonary hypertension.121 Lesions in the left atrium that may cause chronic pulmonary venous hypertension include cor triatriatum and myxoma. Myxoma of the left atrium frequently produces obstruction to blood flow. Clinically, it may mimic mitral stenosis. Chronic pulmonary venous hypertension may be caused by rheumatic mitral stenosis. This is often accompanied by a degree of mitral incompetence, although either condition may occur in isolation. Less common causes of mitral incompetence include infective endocarditis, mucoid degeneration of the mitral valve, and ischemic heart disease causing rupture of the chordae tendineae or papillary muscle dysfunction. In infants and children, congenital mitral stenosis or atresia may produce pulmonary venous hypertension, and congestive vasculopathy may already be present at birth. Left ventricular failure is a common cause of pulmonary venous hypertension. Causes include ischemic heart disease, systemic hypertension, myocarditis, and cardiomyopathy. Finally, pulmonary venous hypertension may result from congenital and acquired lesions of the aortic valve, and from coarctation of the aorta.
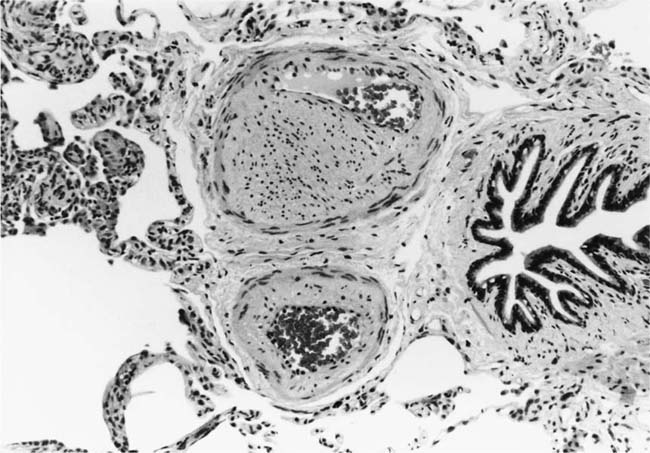
FIGURE 25–26 Open lung biopsy from a patient with thrombotic obstruction of the distal pulmonary arteries. The muscular pulmonary artery in the upper half of the picture shows a cushionlike eccentric patch of intimal fibrosis. The muscular pulmonary artery in the lower half of the picture shows concentric intimal fibrosis (H&E, ×200).
Pulmonary Arteries and Arterioles
The pulmonary arterioles become muscularized with the development of a distinct tunica media of circular smooth muscle sandwiched between internal and external elastic laminae.120,122 The muscular pulmonary arteries show medial hypertrophy, which is frequently accompanied by intimal fibrosis and adventitial fibrosis.119,120,122 In contrast to other forms of pulmonary hypertension, in pulmonary venous hypertension there is a regional distribution of the vascular lesions. In patients with mitral stenosis, the medial hypertrophy of muscular pulmonary arteries is more pronounced at the bases of the lungs than at the apices.123 On the other hand, intimal fibrosis is often more conspicuous in the upper parts of the lungs.
Medial Hypertrophy
In patients with chronic pulmonary venous hypertension, medial hypertrophy of muscular pulmonary arteries is common and often severe (Fig. 25–31). In mitral stenosis, the increase in medial thickness may be greater than that seen in patients with ventricular septal defect. In adults with mitral stenosis, there is a poor correlation between the medial thickness, and the pulmonary artery pressure and severity of mitral valve disease.124,125 There may be severe medial hypertrophy with only mild elevation of the pulmonary artery pressure. This discrepancy may be because some patients with mitral valve disease may have a normal pulmonary artery pressure at rest but develop significant pulmonary hypertension on exercise. Intermittent elevation of the pulmonary artery pressure may be associated with considerable medial hypertrophy. Another factor may be interstitial edema causing thickening of the arterial wall. An increase in the number of collagen fibers between medial smooth muscle cells can be seen by light microscopy, and ultrastructural studies have shown an increase in the amount of collagen in the tunica media of patients with mitral stenosis.125,126 This interstitial edema and fibrosis explains why adults with mitral stenosis have a reduced number of smooth muscle nuclei per unit medial area of pulmonary arteries compared with controls.125 Unlike adults, children with mitral stenosis have the same density of medial smooth muscle nuclei as normal children,125 and there is rough correlation between the medial thickness of muscular pulmonary arteries and the pulmonary artery pressure.
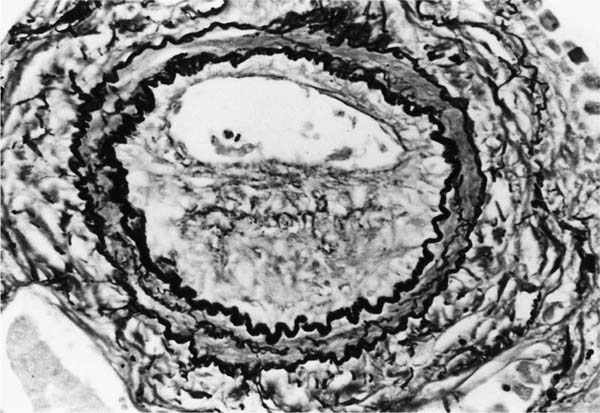
FIGURE 25–27 Muscular pulmonary artery showing eccentric intimal fibrosis. From a patient with thrombotic obstruction of the distal pulmonary arteries (EVG, ×500).
Intimal Fibrosis
Cellular intimal proliferation is uncommon in patients with chronic pulmonary venous hypertension but intimal fibrosis extending over long distances is very common. It is usually eccentric, but even when it is concentric it does not have the laminar and onion-skin pattern that is characteristic of hypertensive pulmonary vascular disease complicating systemic-to-pulmonary shunts. The intimal fibrosis may be severe, but obliteration of the lumen is rare. The pathogenesis of the intimal fibrosis that occurs in chronic pulmonary venous hypertension is not clear.120 It may result from the organization of thrombus, but interstitial edema may also play a role.123
Other Arterial Lesions
Longitudinal smooth muscle cells may be intermingled with the intimal fibrosis in small muscular pulmonary arteries. Longitudinal muscle may also be encountered in the tunica media and adventitia, adjacent to the internal elastic lamina, and external elastic lamina, respectively.51 Interstitial edema may play a role in the development of the adventitial fibrosis (Fig. 25–31).127 Fibrinoid necrosis and arteritis have been described in the muscular pulmonary arteries of patients with mitral stenosis, but these lesions are very rare.118,128 Plexiform lesions do not occur in uncomplicated chronic pulmonary venous hypertension. Occasionally they have been found in children with congenital mitral stenosis or atresia, but these cases have always been complicated by an additional defect associated with a systemic-to-pulmonary shunt.63 The advanced medial hypertrophy that occurs in chronic pulmonary venous hypertension may protect the muscular pulmonary arteries from the development of fibrinoid necrosis, arteritis, and plexiform lesions.27
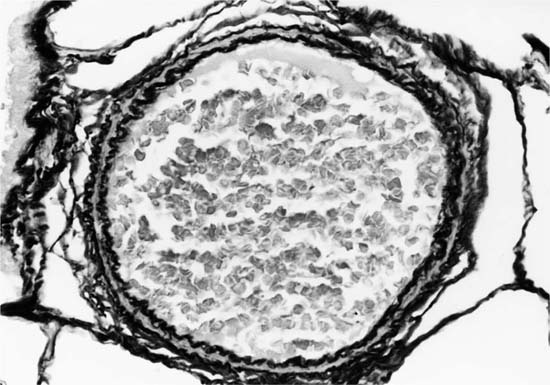
FIGURE 25–28 Muscular pulmonary artery from the patient with thrombotic obstruction of the distal pulmonary arteries illustrated in Fig. 25–26. The vessel is devoid of medial hypertrophy and intimal fibrosis (EVG, ×500).
Alveolar Capillaries
In histologic sections, the alveolar capillaries may be prominent and dilated but not necessarily engorged with blood. Ultrastructural studies of lung biopsy specimens derived from patients with mitral stenosis have revealed swelling and edema of the capillary endothelial cells that develop vacuoles (Fig. 25–32).129 The swollen endothelial cytoplasm may project into the capillary lumen. The basal lamina of the endothelial cells becomes thickened, particularly on the septal aspect of the capillary wall.59,129 Occasionally, the endothelial cell basal lamina is split to enclose disintegrating fragments of extravasated erythrocytes (Fig. 25–33). Proliferated collagen surrounds some of the alveolar capillaries and appears to displace them inward from their normal superficial position in the alveolar wall.129,130
Pulmonary Veins
In chronic pulmonary venous hypertension the pulmonary veins show intimal fibrosis, medial hypertrophy, and adventitial fibrosis.120 They also show a change referred to as arterialization, in which a distinct tunica media of circular smooth muscle develops between internal and external elastic laminae (Fig. 25–34).119,120,123 Intimal fibrosis is common in the pulmonary veins and venules but it is usually not severe and never obstructive (Fig. 25–35). It is acellular and resembles that due to age change. However, it is more pronounced than that seen as an age change in normal adults. Medial hypertrophy and arterialization of pulmonary veins are much more prominent at the base of the lung than at the apex.123 This is in contrast to the intimal fibrosis, which is more marked at the apex than at the base, a situation resembling that of the pulmonary arteries.
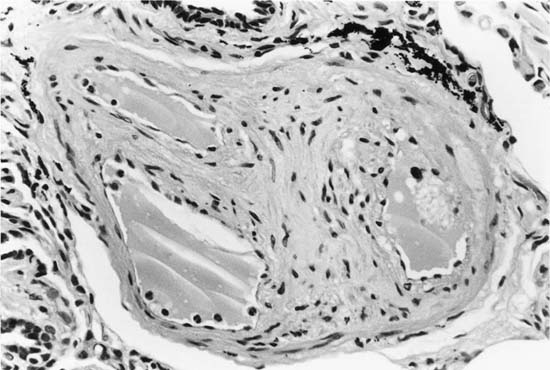
FIGURE 25–29 Muscular pulmonary artery showing a colander-like lesion resulting from organization of thrombus. The recanalized lumen is represented by three vascular channels. From the patient illustrated in Fig. 25–26 (H&E ×500).
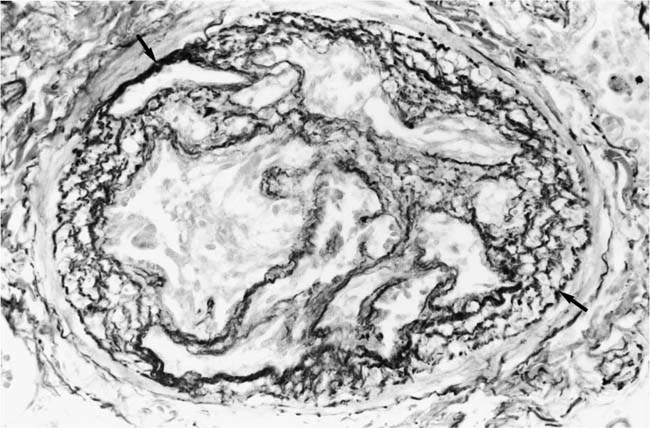
FIGURE 25–30 The lumen of this muscular pulmonary artery contains multiple fibroelastic septa resulting from organization of thrombus. Note that the internal elastic lamina (two arrows) is intact (EVG, ×500).
Pulmonary arteries |
Medial hypertrophy |
Muscularization of arterioles |
Intimal fibrosis |
Adventitial fibrosis |
Arteritis (rare) |
Alveolar capillaries |
Dilatation and congestion |
Endothelial cell swelling |
Thickening of basement membrane |
Extravasation of erythrocytes |
Pulmonary veins |
Medial hypertrophy |
Arterialization |
Intimal fibrosis |
Lymphatics |
Dilatation |
Interstitial tissue and alveoli |
Interstitial edema |
Interstitial fibrosis |
Proliferation of type II pneumonocytes |
Hemorrhage |
Hemosiderosis |
Mast cell proliferation |
Osseous metaplasia |
Alveolar microlithiasis |
Pulmonary veins |
Pulmonary veno-occlusive disease |
Pulmonary capillary hemangiomatosis |
Congenital stenosis of main pulmonary veins |
Congenital stenosis of left venoatrial junction |
Anomalous pulmonary venous drainage |
Left atrium |
Cor triatriatum |
Myxoma of left atrium |
Mitral valve |
Congenital stenosis or atresia |
Rheumatic stenosis or incompetence |
Incompetence due to endocarditis, and so forth |
Left ventricle |
Failure due to ischemia, hypertension, myocarditis, and so forth |
Aortic valve |
Congenital stenosis or atresia |
Acquired stenosis or incompetence |
Aorta |
Coarctation |
Mediastinum |
Fibrosing mediastinitis |
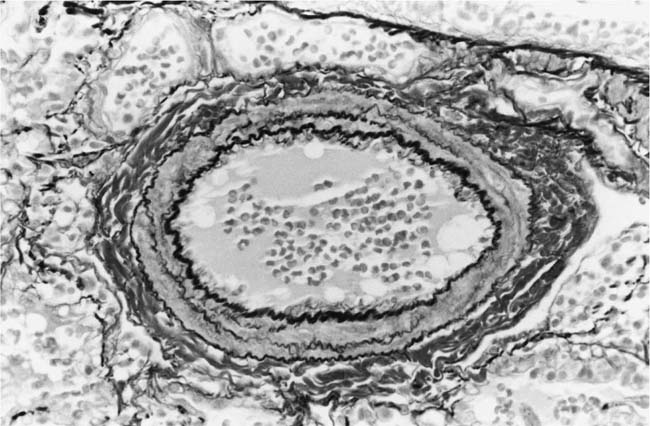
FIGURE 25–31 Muscular pulmonary artery showing medial hypertrophy and adventitial fibrosis. Open lung biopsy from a 21-year-old man with chronic pulmonary venous hypertension secondary to left ventricular failure due to systemic hypertension associated with chronic glomerulonephritis (EVG, ×500).
FIGURE 25–32 Alveolar-capillary wall in a 43-year-old woman with mitral stenosis. Lucent areas of edema (I) separate septal cells (S) and connective tissue fibrils (F). The granular amorphous zone (Z), which separates the membranous pneumonocyte (M) from the edematous endothelial cell (E) over the convexity of the capillary, is not edematous [electron micrograph (EM), ×25,025]. (From Kay and Edwards,129 with permission.)
Lymphatics, Interstitial Tissue, and Airways
Pulmonary edema is a significant part of the pathophysiology of chronic pulmonary venous hypertension. Histologically, there is dilatation of lymphatic channels and widening of the interlobular fibrous septa due to interstitial edema (Fig. 25–36). Ultrastructural studies of open lung biopsy specimens from patients with mitral stenosis show that interstitial edema is confined to the thicker collagen-containing portions of the alveolar wall, and does not involve the thin portions of the blood–air barrier that are devoid of collagen and situated over the convexities of the alveolar capillaries (Fig. 25–32).129 Alveolar edema is a late and not inevitable manifestation of pulmonary edema. The permeability of the interendothelial junctions of the alveolar capillaries appears to be dependent on intraluminal pressure. Alveolar epithelium rather than capillary endothelium seems to be the critical barrier to the entry of water and solutes into the alveolar spaces.
Chronic edema leads to interstitial fibrosis118 associated with a proliferation of mast cells in the lung.277 The alveolar capillaries become embedded in dense connective tissue. The overlying alveoli are lined by type II pneumonocytes corresponding to the “cuboidal cell metaplasia” seen on light microscopy.118,129 It is possible that the type II cells have replaced type I pneumonocytes damaged by stress failure.131 The interstitial fibrosis may be associated with hemosiderosis. Hemosiderin, derived from erythrocytes, is present in alveolar macrophages (Fig. 25–37) or deposited within the interstitium, particularly in relation to blood vessels, bronchi, and the interlobular fibrous septa. Occasionally, hemosiderin encrusts elastic fibers in the pulmonary blood vessels and alveolar septa where it may provoke a giant cell reaction. In some instances this has led to the inappropriate diagnosis of “endogenous pneumoconiosis.”132 Ossification may occur in the alveolar spaces in chronic pulmonary venous hypertension (Fig. 25–38).119 The metaplastic nodules of lamellar bone are presumed to develop in relation to persistent exudate in the alveolar ducts and spaces. Extensive ossification of the lungs has occasionally been reported in mitral valve disease.133 A rare complication of chronic pulmonary venous hypertension is alveolar microlithiasis, in which uniform stratified concretions of calcium phosphate develop within the alveoli.134 These render the lungs stony hard.
FIGURE 25–33 Alveolar wall in a 45-year-old woman with mitral stenosis. The basement membrane (B) of the endothelial cell (c) lining the capillary is thickened and shows multiple layers. It encloses an extravasated erythrocyte (e). The alveolar space (A) is lined by a membranous pneumonocyte (arrow) (EM, ×12,500).
Relation of Pathologic Changes to Clinical Features
In chronic pulmonary venous hypertension, medial hypertrophy of muscular pulmonary arteries and pulmonary veins is most pronounced at the bases of the lungs, which correlates with the greater pulmonary vascular resistance at the bases and the consequent redistribution of blood flow toward the apices. Postmortem arteriograms show dilatation of the pulmonary arteries or no change in their size in the upper parts of the lungs, whereas those in the lower parts are narrowed.135 The presence of dilated lymphatic channels within edematous, thickened interlobular fibrous septa (Fig. 25–36) is the morphologic basis of the transient basal horizontal (Kerley B) lines that are visible in the chest radiograph when the mean left atrial pressure exceeds 24 mm Hg (Fig. 25–39).136 The degree of distention of the pulmonary lymphatics has been assessed in lung biopsy specimens from patients with mitral stenosis.137 Clusters of obviously distended lymphatics around pulmonary arteries were observed in 7 of 20 cases of mitral stenosis, and in 6 of these the pulmonary artery wedge pressure was 30 mm Hg or more. Pulmonary congestion, edema, and dilatation of lymphatics are particularly prominent in the upper parts of the lung.123 Patients with mitral stenosis and chronic left heart failure have a surprising resistance to pulmonary edema despite persistently increased pulmonary venous pressure, and pulmonary edema may only appear terminally or as a complication of fast atrial fibrillation. It is possible that this resistance to pulmonary edema may be attributable in part to a decrease in pulmonary microvascular permeability.138,139 Chronic edema leads to interstitial fibrosis of the lung, which has a haphazard distribution. The combination of interstitial fibrosis and hemosiderosis is responsible for the characteristic appearance of brown induration of the lungs at autopsy.
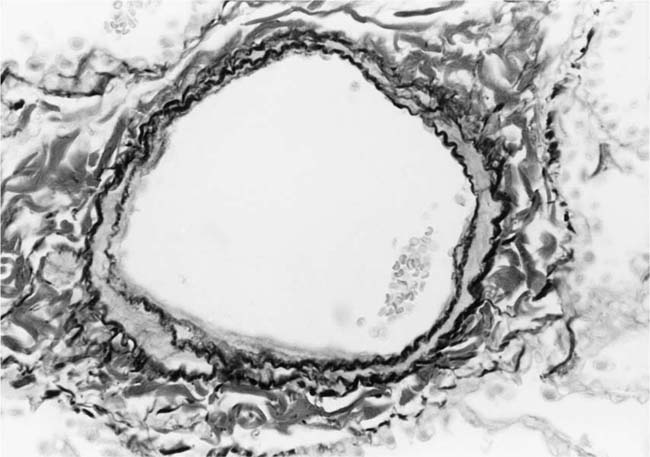
FIGURE 25–34 Pulmonary vein from the patient illustrated in Fig. 25–31. The vessel is located in an interlobular fibrous septum. The wall shows arterialization. A distinct tunica media of circular smooth muscle has developed between internal and external elastic laminae (EVG, ×500).
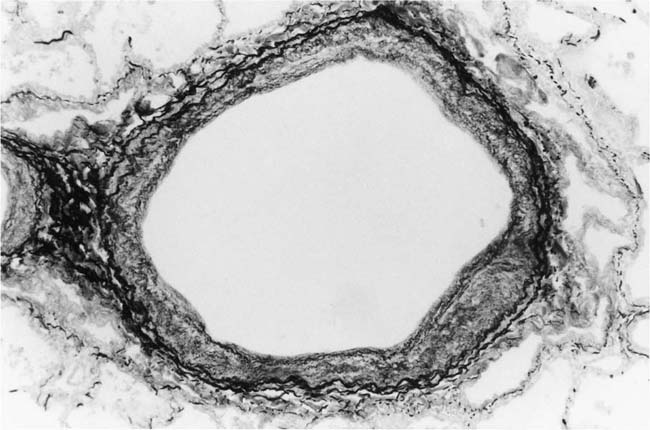
FIGURE 25–35 Pulmonary vein showing intimal fibrosis. It is from a 21-year-old woman with chronic pulmonary venous hypertension secondary to left ventricular failure. She suffered from diabetes mellitus and systemic hypertension (EVG, ×200).
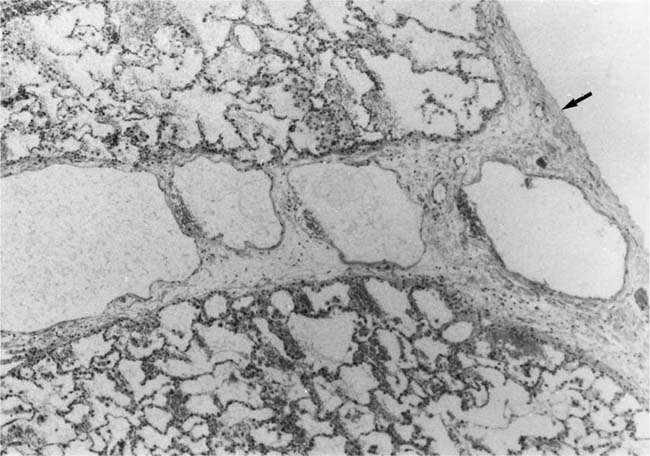
FIGURE 25–36 Dilated lymphatic channel in an edematous interlobular fibrous septum. Note the pleura (arrow) and the congested alveolar capillaries particularly in the lower half of the picture. From the patient with chronic pulmonary venous hypertension illustrated in Figs. 25–31 and 25–34. His chest radiograph showed basal horizontal (Kerley B) lines (EVG ×80).
Recurrent pulmonary hemorrhage with associated hemoptysis and hemosiderosis is a characteristic feature of chronic pulmonary venous hypertension.58,140 Early in the course of the disease, when the pulmonary vascular resistance is relatively low, the hemorrhage is probably due to stress failure of the pulmonary capillaries. The walls of the alveolar capillaries are extremely thin to allow respiratory gases to diffuse through them easily. When the capillary blood pressure is raised, damage to the wall occurs, resulting in stress failure with subsequent pulmonary hemorrhage.131,140 The pressure at which stress failure occurs in human capillaries is not known. However, it occurs at a microvascular pressure of 40 mm Hg in rabbits and 70 mm Hg in dogs.140 The strength of the thin part of the alveolar wall can be attributed to type IV collagen in the extracellular matrix.140 In chronic pulmonary venous hypertension, the collagen content of the alveolar wall increases, and the capillary basement membrane thickens (Fig. 25–33), making it more resistant to rupture.129,130 It has been suggested that in patients with established mitral stenosis, much of the bleeding is from bronchial submucosal veins that are exposed to high pressure through anastomoses with the pulmonary veins, rather than from the alveolar capillaries.52,140 There is no direct relation between the occurrence and severity of pulmonary hemosiderosis and the blood pressure in either the pulmonary arteries or pulmonary veins.58
Chronic pulmonary venous hypertension is associated with impairment of gas exchange across the alveolar-capillary wall.141 It is difficult to correlate structural changes in the lung in mitral stenosis with respiratory function because the histologic changes tend to be patchy, and vary in different parts of the lung. In one study of lingula biopsy specimens from patients with mitral stenosis, there was a poor relation between structural abnormalities in alveolar walls and respiratory function tests.124 Diffusing capacity, vital capacity, and maximum breathing capacity were not related to the presence or degree of fibrosis of alveolar walls. The impairment of diffusing capacity seemed to depend less on the structure of the alveolar wall than on the presence of occlusive pulmonary vascular changes.
FIGURE 25–37 Hemosiderosis in chronic pulmonary venous hypertension. The alveolar spaces contain large clumps of hemosiderin-laden macrophages. There is congestion of the alveolar capillaries. From the patient illustrated in Fig. 25–35 (Perls Prussian blue stain with H&E, ×200).
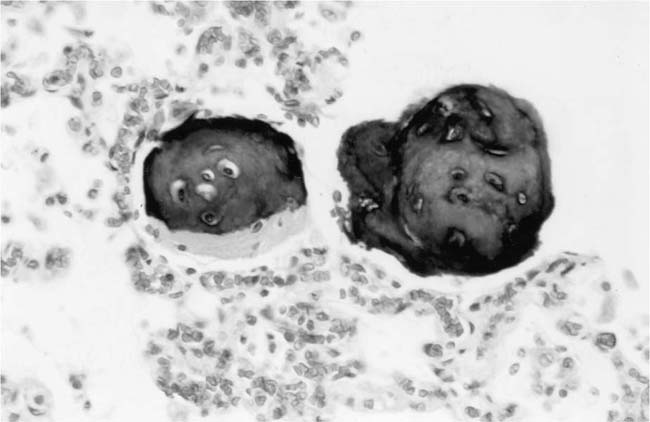
FIGURE 25–38 Bone metaplasia in chronic pulmonary venous hypertension. The alveolar spaces of this 17-year-old boy with pulmonary veno-occlusive disease contain two nodules of lamellar bone (H&E ×500).
FIGURE 25–39 Basal horizontal (Kerley B) lines extend to the periphery of the lung in the right lower zone of this chest radiograph from a patient with pulmonary edema.
Reversibility of Pulmonary Hypertension and Vascular Lesions
Chronic pulmonary venous hypertension is potentially reversible,130 and in most patients with mitral stenosis the elevated pulmonary vascular resistance falls rapidly almost to normal within 24 hours of mitral valve surgery.10 It is rare for a pathologist to be able to compare the pulmonary vascular lesions at lung biopsy during mitral valve surgery with the situation in the same patient many years later. Consequently, published studies of the regression of pulmonary vascular changes following mitral valve surgery tend to be anecdotal.
In one case, the original lung biopsy revealed medial hypertrophy and mild intimal fibrosis of pulmonary arteries. Eleven years later the medial thickness was near normal and the intimal fibrosis had disappeared.142 In another case, the lung biopsy taken at the time of mitral valve surgery showed pronounced medial hypertrophy and intimal fibrosis of pulmonary arteries. The pulmonary veins showed medial hypertrophy, arterialization, and intimal fibrosis.143 Ten years later, when the patient died as a result of an accident, the pulmonary arteries showed minimal medial hypertrophy and intimal fibrosis. The pulmonary veins were normal except for some residual intimal fibrosis. The few patients with mitral stenosis who die after surgery with persistently elevated pulmonary arterial pressure seem to have sufficiently severe pulmonary vascular disease to account for their persistent pulmonary hypertension and death.144
Pulmonary veins and venules |
Obstructive intimal fibrosis |
Recanalized thrombus |
Intravascular fibrous septa |
Medial hypertrophy |
Arterialization |
Adventitial fibrosis |
Nonspecific phlebitis (rare) |
Granulomatous phlebitis (rare) |
Alveolar capillaries |
Dilatation and congestion |
Thickening of basement membrane |
Extravasation of erythrocytes |
Pulmonary arteries |
Intimal fibrosis |
Medial hypertrophy |
Recanalized thrombus |
Intravascular fibrous septa |
Recent thrombus |
Adventitial fibrosis |
Arteritis (rare) |
Lymphatics |
Dilatation |
Interstitial tissue and alveoli |
Interstitial edema |
Interstitial fibrosis |
Proliferation of type II pneumonocytes |
Intra-alveolar hemorrhage |
Hemosiderosis |
Osseous metaplasia |
Pulmonary Veno-occlusive Disease
Höra145 published in 1934 the first well-documented case of pulmonary veno-occlusive disease. Subsequently, several cases were reported under a variety of terms until pulmonary veno-occlusive disease became the generally accepted term in 1966.146 Approximately 150 well-documented cases have now been reported in the literature, with the largest series comprising 11 cases.147 Pulmonary veno-occlusive disease affects all age groups and has been reported from all over the world. About 60% of patients are male and the age range is from 11 days to 76 years, with ~40% of patients being age 16 years or less. One case was presumed to be of intrauterine origin.148 The predominant lesions are in the pulmonary veins but there are also significant changes in the alveolar capillaries, muscular pulmonary arteries, lymphatics, and lung parenchyma (Table 25–10).
Pulmonary Veins
The intrapulmonary veins and venules are narrowed (Fig. 25–40) or occluded (Fig. 25–41) by intimal fibrosis,146,149 which may consist of either loose, myxoid connective tissue, or dense collagen and elastic tissue sometimes including occasional smooth muscle cells.150 The intimal fibrosis may be eccentric (Fig. 25–40) or concentric (Fig. 25–41), with the concentric form affecting mainly small veins less than 100 μm in diameter.120 Colander lesions (Fig. 25–42) and intravascular fibrous septa indicative of recanalization are present in pulmonary veins in most cases, but recent thrombus is identified in only ~25% of patients.151 In most cases both the pulmonary veins in the interlobular fibrous septa and the intraacinar veins and venules are involved. However, in some patients the lesions are virtually restricted to the small intraacinar veins and venules, which makes histologic diagnosis difficult because of the problem of distinguishing small pulmonary veins and venules from pulmonary arterioles. The proportion of veins and venules affected varies between 30 and 90%.149 In most cases the venous changes are equally and evenly distributed in both lungs. In a few cases, however, the veins in some parts of the lungs are more affected than in others. There is no constant preference for specific areas of the lung. The media of the pulmonary veins often shows hypertrophy and arterialization with the development of distinct internal and external elastics laminae (Figs. 25–40 and 25–43). In some cases, these features are probably secondary to increased pressure in the pulmonary veins resulting from obstruction in the larger distal venous trunks (Fig. 25–44). In other cases, where the larger veins are widely patent, the medial hypertrophy and arterialization may result from direct injury to the walls.151 Pulmonary veins of all sizes frequently show thickening of the adventitia due to deposition of collagen and elastic tissue (Fig. 25–44).120 The lesions in the pulmonary veins are usually bland, but occasional cases of nonspecific pulmonary phlebitis (Fig. 25–45)152 and granulomatous pulmonary phlebitis (Fig. 25–46)153 have been described. Ultrastructural examination of the small pulmonary veins has shown thickening of the wall due to a haphazard arrangement of smooth muscle cells, collagen, and extracellular matrix.150
Alveolar Capillaries
The alveolar capillaries may show dilatation and severe congestion particularly in relation to occluded or narrowed pulmonary venules. Electron microscopy of alveolar walls has been reported in 10 cases. In eight cases it was negative. In two cases, electron-dense deposits were identified. In one of these cases, immunofluorescent microscopy demonstrated deposits of immunoglobulin G (IgG) and complement in the alveolar walls, so the electron-dense deposits were interpreted as immune complexes.154 In the other case, immunofluorescent microscopy was negative and it was considered that the electron-dense deposits represented fragments of disintegrating extravasated erythrocytes (Fig. 25–47).150 In this case, the endothelial cell basement membrane was also thickened.
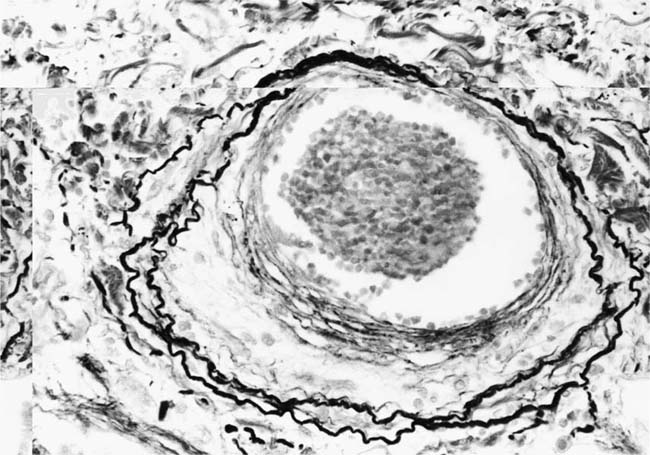
FIGURE 25–40 Pulmonary vein from a 47-year-old woman with pulmonary veno-occlusive disease. There is eccentric intimal fibrosis with narrowing of the lumen. In the lower half of the picture, the wall of the vessel shows arterialization. A distinct media of smooth muscle has developed between an internal and external elastic (EVG, ×500).
Muscular Pulmonary Arteries
In the majority of cases of pulmonary veno-occlusive disease, the muscular pulmonary arteries show intimal fibrosis, medial hypertrophy (Fig. 25–48), and adventitial fibrosis.120 In 75% of cases there is eccentric intimal fibrosis (Fig. 25–48), which appears to be postthrombotic in origin and thus differs from the intimal fibrosis that occurs in mitral stenosis.151 Intravascular fibrous septa and recent thrombi are encountered in the pulmonary arteries in 25% of cases (Fig. 25–49). Plexiform lesions and dilatation lesions have not been identified, but pulmonary arteritis (Fig. 25–50) has occasionally been observed.155
Lymphatic Channels
The lymphatic channels in the interlobular fibrous septa, perivascular (Figs. 25–1 and 25–42), and peribronchiolar connective tissue are frequently dilated. The presence of dilated lymphatic channels within thickened and edematous interlobular fibrous septa (Fig. 25–36) is the morphologic basis of the Kerley B lines (Fig. 25–39) that are observed in ~20% of cases of pulmonary veno-occlusive disease.
Lung Parenchyma
Interstitial edema is common and is most easily recognized in the interlobular fibrous septa, which become abnormally wide (Fig. 25–36). In long-standing cases, interstitial fibrosis, which may be severe and extensive, develops in the alveolar walls.149 A proliferation of type II pneumonocytes150 and an infiltrate of lymphocytes and plasma cells frequently accompanies the fibrosis, so that the histology mimics usual interstitial pneumonia (Fig. 25–51).149 Hemosiderin, derived from extravasated erythrocytes, is present in alveolar macrophages or deposited within the interstitium. Occasionally, hemosiderin encrusts elastic fibers in the walls of pulmonary blood vessels (Fig. 25–52) and alveolar septa, where it may evoke a giant cell reaction. The hemosiderosis may be so prominent that a diagnosis of idiopathic pulmonary hemosiderosis is entertained. Nodules of metaplastic bone may occur within the alveolar spaces.150 Pulmonary infarction has been observed in a few cases.
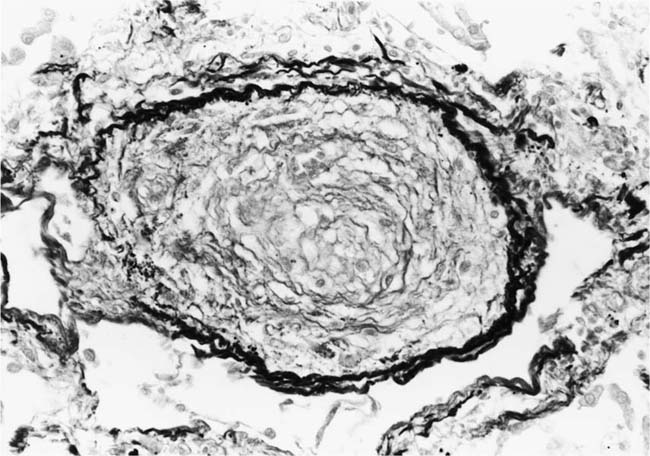
FIGURE 25–41 Pulmonary veno-occlusive disease. The lumen of this small pulmonary vein is totally occluded by loose connective tissue. Note the dilated perivascular lymphatic at the bottom of the picture. Same case as Fig. 25–40 (EVG, ×500).
Hemodynamics and Prognosis
One hemodynamic observation that is described by many investigators of pulmonary veno-occlusive disease is the failure to obtain a pulmonary artery wedge pressure tracing. When the catheter is wedged in a pulmonary artery and it is flushed, the pressure rises disproportionately and falls extremely slowly to baseline because the infused saline becomes trapped between the catheter tip and the narrowed pulmonary veins. If the pulmonary artery catheter is successfully wedged, the pulmonary artery wedge pressure obtained is usually normal or decreased, despite the fact that the pulmonary capillary pressure is elevated. This is because in the wedged position, the catheter abuts a static column of blood that extends beyond the pulmonary venules to the central pulmonary veins and the left atrium.156,157 The left atrial pressure, which is usually normal in pulmonary venoocclusive disease, largely determines the pulmonary artery wedge pressure.
Sixty-eight well-documented cases of pulmonary venoocclusive disease were published between 1934 and 1986, and the hemodynamic findings were given in 40. The pulmonary artery systolic pressure ranged from 30 to 135 mm Hg with a mean value of 85 mm Hg (n = 29). The mean pulmonary artery pressure was mentioned in 19 reports: it ranged from 28 to 107 mm Hg, with a mean of 64 mm Hg. Elevation of the pulmonary artery wedge pressure to values greater than 12 mm Hg was recorded in 14 of 34 cases, the highest mean value being 25 mm Hg. The duration of the disease from onset of symptoms to death was recorded in 47 of the 68 case reports. It ranged from 1 to 84 months. In 37 cases (79%), the duration was less than 2 years.
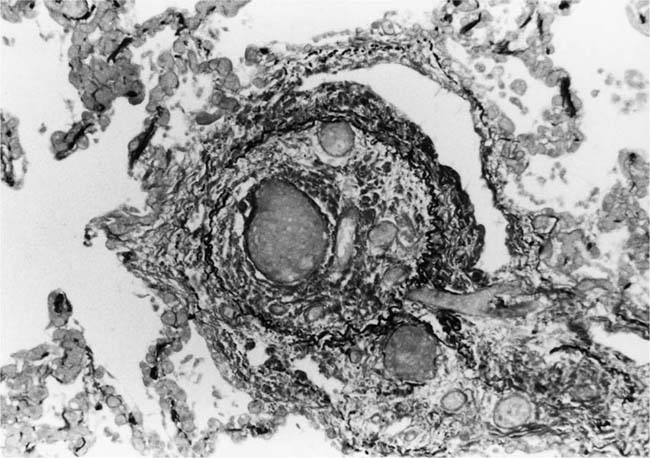
FIGURE 25–42 Pulmonary veno-occlusive disease. Pulmonary vein showing colander lesion consistent with old organized and recanalized thrombus. Note the congestion of the surrounding alveolar capillaries and the dilated perivascular lymphatic in the right upper quadrant. Same case as Fig. 25–40 (EVG, ×200).
In a retrospective pathologic study that included 25 cases of pulmonary veno-occlusive disease, the mean pulmonary artery pressure was 60 mm Hg and the mean survival time 14 months.79 In five cases of pulmonary veno-occlusive entered in the National Heart, Lung, and Blood Institute Registry of Primary Pulmonary Hypertension, the mean pulmonary artery pressure (± SD) was 61 ± 11 mm Hg; the mean and median survival times were 289 and 84 days, respectively.77
Etiology
The cause of pulmonary veno-occlusive disease is unknown but the character of the venous lesions indicates that thrombosis is an essential factor in the vast majority of cases, if not in all. Changes in the pulmonary arteries have attracted less attention than those in the pulmonary veins despite the fact that they are sometimes as severe.151 This suggests that the process is a pulmonary vascular occlusive disease rather than a condition limited to the pulmonary veins. In some cases, the arterial lesions appear to be more recent than those in the veins. This suggests that the process may be begin in the pulmonary veins and that the arteries are affected later. It may be that the pulmonary veins are more susceptible to injury than the pulmonary arteries. Normal pulmonary venous endothelium contains less plasminogen activator than pulmonary arterial endothelium. If an infective or toxic agent damaged the pulmonary vascular endothelium, plasminogen activator might be depleted in the pulmonary veins before the pulmonary arteries with the subsequent encouragement of thrombosis in the pulmonary veins. Studies of blood coagulation in patients with pulmonary veno-occlusive disease are rare. In one case the platelet adhesiveness was increased and platelet survival was short. The fibrinolytic system was normal.158
A wide variety of risk factors and associations of pulmonary veno-occlusive have been described (Table 25–11). Most of these are based on reports of single cases and small series of cases. It is unlikely that a single etiologic agent is responsible for all cases of pulmonary veno-occlusive disease. It is probably best regarded as a syndrome rather than an etiologic entity, although a final common pathway, probably thrombosis, is likely.
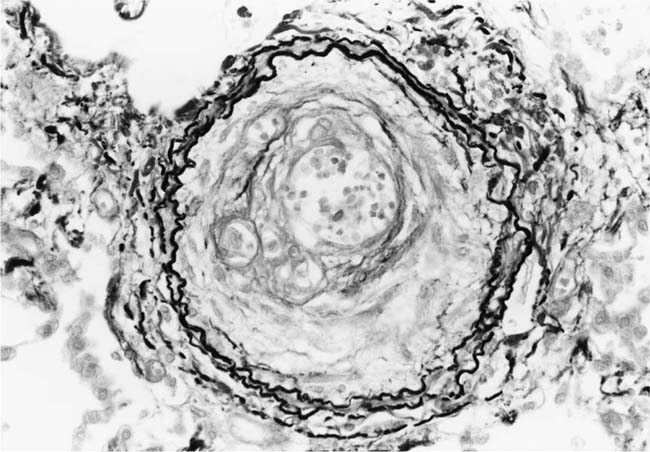
FIGURE 25–43 Pulmonary veno-occlusive disease. This pulmonary vein shows intimal fibrosis. The lumen is narrowed by loose connective tissue including multiple small vascular channels consistent with recanalized thrombus. The wall shows arterialization. A medial coat of smooth muscle is bounded by internal and external elastic laminae. From the case of the case of pulmonary veno-occlusive disease illustrated in Fig. 25–40 (EVG, ×500).
Infection
It has been suggested that thrombosis may be initiated by a viral infection because of the frequent concomitant histologic finding of mild interstitial pneumonia, and a history of a preceding influenza-like illness in many cases.149,157 However, no specific virus has been implicated, and it has been postulated that the interstitial disease and respiratory symptoms might be the consequence rather than the cause of the pulmonary venous occlusion.149 Serologic evidence suggestive of recent infection with one of several agents including Toxoplasma gondii and measles has been documented around the time when pulmonary veno-occlusive disease was diagnosed.146,158 Other patients have displayed features suggestive of Epstein-Barr virus or cytomegalovirus infection around the time of diagnosis of pulmonary veno-occlusive disease.152
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


