Pulmonary Lymphangioleiomyomatosis
Lymphangioleiomyomatosis (LAM) is a multisystem disorder, predominantly affecting women, which is characterized by cystic lung lesions, abdominal angiomyolipomas (AML) and lymphatic abnormalities, for example, lymphatic tumors, chylous effusions.1–5 These pathologic features are caused by the proliferation of a neoplastic smooth muscle–like LAM cell that also has characteristics of melanocytes.6 Inherited and sporadic forms of LAM have been described. Sporadic LAM is caused by somatic mutations in an unknown susceptible cell of the tuberous sclerosis complex (TSC) 2 (TSC2) gene.7,8 LAM also occurs in TSC, an autosomal dominant disorder resulting from germline mutations in the TSC1 or TSC2 genes that is characterized by widespread hamartomas in several organs including the brain, heart, skin, kidney, eyes, lung, and liver, and occurs in 1 of 6000 live births.9
EPIDEMIOLOGY
Until the establishment of LAM registries,1 LAM was considered to be a fatal disease of women of child-bearing age for which oophorectomy, antiestrogen therapy, and lung transplantation were the only therapeutic options.10–13 LAM is now best defined as a chronic disease of post- and premenopausal women with a life expectancy spanning decades.14 Sporadic LAM is an uncommon disease occurring in approximately 4.9/1,000,000 women.15 Although the association of TSC and cystic lung disease has long been recognized,11–13 little is known about the prevalence and the natural history of LAM in TSC (TSC-LAM). The prevalence of cystic lung disease in women with TSC was reported to range from 30% to 40%; in male patients, it has been estimated to be 13%.16 Males with TSC tend to have milder, subclinical lung involvement.
CLINICAL PRESENTATION
Patients with LAM often present with a history of progressive dyspnea. Pneumothorax, another common presentation of LAM, is often recurrent, occurring in about 50% to 60% of patients.1–4 The size of the lung cysts, as seen on high-resolution computed tomography (HRCT) scans (Fig. 62-1A), appears to parallel the incidence of pneumothorax; higher incidence of pneumothorax is seen in patients with larger cysts.17 Other modes of presentation include chylothorax, abdominal lymphangioleiomyomas, chylous ascites, hemoptysis, chyluria, chyloptysis, and hemorrhage caused by renal AML (Table 62-1).1,5,18 Lymphatic involvement in LAM occurs in the posterior mediastinum, retroperitoneal and pelvic areas and includes lymphadenopathy, chylous effusions, and lymphangioleiomyomas.5,18 AML are benign tumors, usually localized in the kidneys and found in approximately 90% of patients with TSC-LAM and 30% of those with sporadic LAM (Table 62-1).1,5,18 The physical examination of LAM patients may disclose wheezing, pleural effusions, ascites, or intra-abdominal masses. In patients with TSC, typical skin lesions or signs of brain involvement may be evident.9
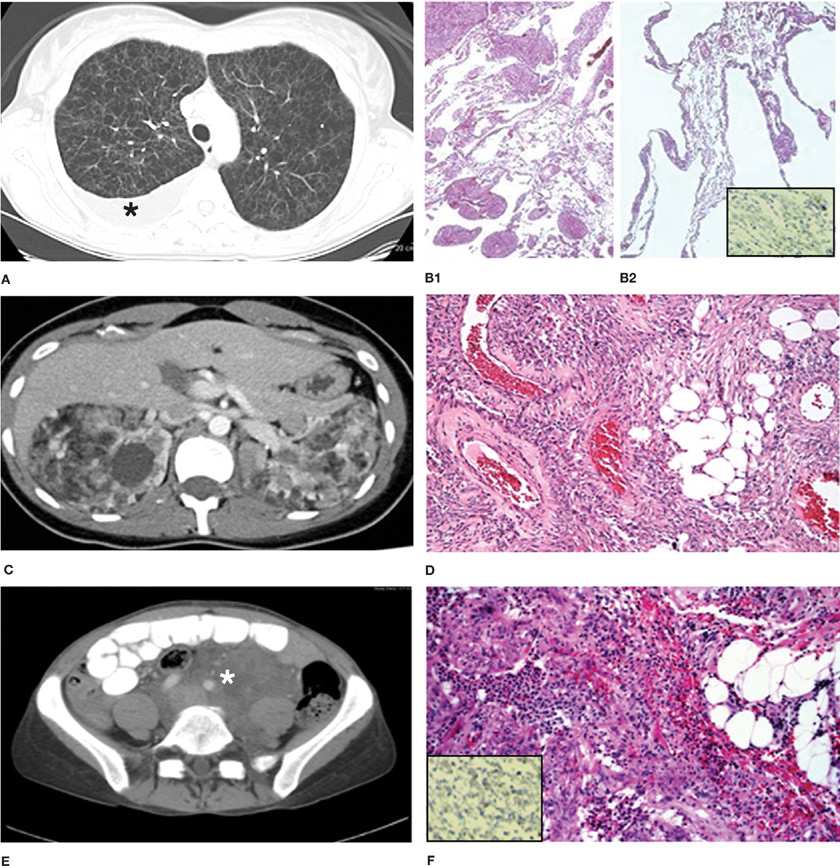
Figure 62-1 Computed tomography scans showing pulmonary and extrapulmonary images of patients with LAM, and the corresponding histopathologic findings. A. Multiple thin-walled cysts scattered throughout the lungs that have completely replaced the normal lung parenchyma. The black asterisk indicates the presence of a right pleural effusion. B1 and B2. Histopathology of the lung showing characteristic nodular smooth muscle cell–like infiltrates and cystic lesions. Inset image on B2 shows immunocytochemistry of lung tissue showing reactivity with monoclonal antibody HMB45. C. Bilateral angiomyolipomas in a patient with TSC-LAM. The fatty, low-density component is clearly visualized. D. Histopathology of AML showing smooth muscle cell–like infiltrates, fatty tissue, and poorly differentiated vascular structures. E. Large, fluid-filled lymphangioleiomyoma (white asterisk) surrounding vascular structures. F. Histologic appearance of a lymphangioleiomyoma showing smooth muscle–like cells arranged in fascicular, trabecular or papillary patterns. Inset image on F shows immunocytochemistry of lymphangioleiomyoma tissue showing reactivity with monoclonal antibody HMB45.

PATHOLOGY
Gross examination of lung sections shows cysts ranging in size from 0.2 to 2 cm.6,19 Microscopically, cysts characterize lung lesions and proliferation of LAM cells in the walls of cysts and along blood vessels, lymphatics, and bronchioles (Fig. 62-1B1,B2), causing airways narrowing, vascular wall thickening, lymphatic disruption, and venous occlusion are observed.6,19 Focal hemosiderosis may be present. Often LAM cells grow in a haphazard, disorderly fashion.6,19 Two types of LAM cells have been described. Small, spindle-shaped cells predominate in the center of the lung nodules; epithelioid cells with large cytoplasm predominate at the periphery.6,19 Both cell types react with antibodies against smooth muscle–cell antigens (e.g., smooth muscle α-actin, vimentin, desmin). The epithelioid cells also react with human melanin black antibody (HMB-45), a monoclonal antibody that recognizes gp100, a premelanosomal protein encoded by the Pmel17 gene (Fig. 62-1B2, inset).6,19 Spindle-shaped LAM cells react with proliferating cell nuclear antigen, indicating that these cells are more proliferative.6 Receptors for estrogen, progesterone, insulin-like growth factors, angiotensin II, hyaluronic acid (CD44), chemokines, and erythropoietin have been identified in LAM cells.20–27
AML are highly vascular tumors comprising smooth muscle–like cells, immature, poorly differentiated blood vessels, and fatty tissue (Fig. 62-1D).6,19 Tumors vary in size from 1 mm to more than 15 cm in diameter.18,19 Smooth muscle–like cells found in AML have the same immunocytochemical properties as lung LAM cells (Fig. 62-1D).6,19 All the major cell types in AML (vascular, fat, smooth muscle) may have mutations in the TSC genes.7,8 The blood supply of AML originates from the renal arteries or aberrant vessels and may completely disrupt normal kidney architecture.18,19 Hilar, mediastinal, and retroperitoneal lymphadenopathy may be seen.18,19 The thoracic duct may be thickened and dilated.19 Lymphangioleiomyomas consist of encapsulated lymphatic masses of varying sizes comprising chyle-filled cysts, with infiltration of LAM cells, displaying the immunoreactivity profile of LAM lung cells, arranged in fascicular, trabecular or papillary patterns and associated with slit-like vascular channels (Fig. 62-1F).6,19
PATHOGENESIS
Sporadic LAM is caused by proliferation of neoplastic LAM cells that have mutations or deletions in the TSC2 (16p13) gene.7,8 Consistent with Knudson’s “two-hit” hypothesis of tumor development,28 loss of heterozygosity of TSC2 has been reported in LAM cells isolated from lung, blood, chyle, urine, and AML from both sporadic LAM and TSC-LAM patients.8,29–31 LAM cells having identical mutations were identified in AML and lungs of the same patient.8,31 LAM cells from patients receiving lung transplantation have been detected in the donor lung, suggesting migration from other sites, such as the kidney or lymphatic system, to the lungs.32,33 The metastatic properties of LAM cells were also demonstrated by the presence of LAM cells in blood, urine, expectorated chyle and pleural, or abdominal chylous fluid of LAM patients.29,30 LAM cell clusters, consisting of LAM cell aggregates covered by lymphatic endothelial cells, which have been proposed to originate from lung LAM lesions, have also been identified in chylous fluid.34 Potential sources of lung LAM cells include AML, the lymphatic system, and the uterus,35 in which case they may originate from abnormal leiomyoma.
A role of estrogens in the pathogenesis of LAM has been suggested by its predominance in premenopausal women, worsening of lung disease during pregnancy or following the administration of estrogens36,37 and the presence of estrogen and progesterone receptors in lung and angiomyolipoma LAM cells.20–22 Estrogens promote the proliferation of TSC-null rat ELT3 leiomyoma–derived cells in vitro, and the growth of subcutaneous tumors in a nude mouse xenograph system.38 Estrogens stimulate the growth of human angiomyolipoma TSC2–/– cells, increase the survival and metastatic properties of TSC2–/– ELT3 cells in mice, and enhance matrix metalloproteinase (MMP)-2 activity of LAM lung-derived cells, thereby promoting cell invasiveness.39–41
The mechanism by which interstitial LAM cell proliferation causes lung cyst formation is unknown.42 It has been proposed that compression of the airways by LAM cells leads to distention of the terminal airspaces and cyst formation.6,19 It has also been proposed that degradation of lung elastic fibers is a major cause of the cystic lesions.42 Matrix metalloproteinases, which play a role in lung remodeling and lymphangiogenesis, are associated with LAM lesions.43 LAM nodules contain MMP2, MMP9, MMP1, and MMP activators (MT1-MMP), and their inhibitors (TIMPs).44,45 Levels of TIMP-3, which inhibits some MMP, were reportedly reduced in LAM lesions.45,46 Compared with normal subjects, serum levels of MMP-9 were higher in patients with LAM,47 suggesting that an imbalance between MMP and their inhibitors may contribute to lung destruction.48 Growth of TSC2-null lesions was associated with an increase in MMP activity and vascular endothelium growth factor D (VEGF-D).48 Elastic fibers were demonstrated in alveoli of mice with TSC2-null lesions48 and human lung LAM nodules show disrupted elastic fibers. The presence of lymphatic spaces in the LAM nodules and strong immunoreactivity towards vascular endothelium growth factor C (VEGF-C), VEGF-D, vascular endothelium growth factor receptor (VEGFR) 3, and podoplanin, markers of lymphatic endothelial cells, led to the hypothesis that disorganized lymphangiogenesis enhances metalloproteinase expression and lung remodeling.48 Some combination of these mechanisms may be the best explanation for the pathogenesis of cystic lung destruction.
PULMONARY PHYSIOLOGY
Airflow obstruction was seen in approximately 61% of patients with sporadic LAM1,49; normal spirometry was present in about 31%. The remaining patients had restrictive disease. By comparison, normal lung function was observed in 53% of patients with TSC-LAM (Table 62-1).1 Increased gas trapping may be also present. The cause of airflow limitation in LAM has been attributed to alveolar destruction,50 but a study of pulmonary mechanics showed that lung elastic recoil was not significantly reduced.51 Instead, upstream airway resistance was increased, suggesting increased airways resistance as the major cause of airflow obstruction.51,52 Reduced diffusing capacity (DLCO) occurred in approximately 60% of sporadic LAM patients.1 Most patients have both decreased FEV1 and DLCO, but some patients have only a reduced DLCO.49 Gas exchange, especially during exercise, is often abnormal (Table 62-2).52,53 An abnormal ventilatory response with excessive minute ventilation and reduced breathing reserve are seen during exercise. Baseline and exercise dead space to tidal volume ratio and alveolar–arterial oxygen difference (A-a/O2) are increased both at rest and during exercise.52 The primary determinants of exercise limitation in LAM are airflow limitation, decreased breathing reserve, dynamic hyperinflation,52–54 and limitation of oxygen transfer due to loss of alveolar capillary surface area. The latter exerts a significant effect upon exercise performance, because the increase in physiologic dead space produces excessive ventilation. The interdependence between airflow obstruction, which produces a decrease in the ventilatory reserve, and cystic lung destruction, which affects gas exchange during exercise, leads to severe impairment in exercise performance.52,53 Pulmonary hypertension may also contribute to reduce oxygen transfer during exercise.55
Decreased ![]() O2max
O2max
Increased ![]() E/
E/![]() CO2 at AT
CO2 at AT
Increased ![]() D/
D/![]() T
T
a![]() O2max, peak oxygen uptake; PaO2, arterial oxygen tension;
O2max, peak oxygen uptake; PaO2, arterial oxygen tension; ![]() E/
E/![]() CO2, ventilatory equivalent for CO2; AT, anaerobic threshold;
CO2, ventilatory equivalent for CO2; AT, anaerobic threshold; ![]() D/
D/![]() T, dead space ventilation ratio; A-a/O2, alveolar arterial oxygen tension difference.
T, dead space ventilation ratio; A-a/O2, alveolar arterial oxygen tension difference.
Source: Data from Crausman RS, Jennings CA, Mortensen RL, et al. Lymphangioleiomyomatosis: the pathophysiology of diminished exercise capacity. Am J Respir Crit Care Med. 1996;153:1368; Taveira-DaSilva AM, Stylianou MP, Hedin CJ, et al. Maximal oxygen uptake and severity of disease in lymphangioleiomyomatosis. Am J Respir Crit Care Med. 2003;168:1427.
RADIOLOGY
Chest radiographic findings in LAM range from being normal, to showing a reticular or nodular irregular shadowing or, in advanced stages, severe cystic changes. Computed tomography (CT) demonstrates diffuse, well-defined, round thin-walled cysts scattered throughout the lungs. Cysts vary in size from a few millimeters to up to 2 cm (Fig. 62-1A).56,57 Additional findings are pleural effusions and lung opacities caused by chyle (Fig. 62-1A). Correlation between the extent of the cystic parenchymal replacement, as measured by high-resolution chest CT (HRCT), and the severity of the disease, as determined by spirometry, DLCO or exercise performance, has been reported.53,57–61 Computer analysis of HRCT can quantify the extent of cystic changes and detect abnormalities in areas adjacent to the cysts that may appear to be radiographically normal60,61; these emphysematous changes were also seen by histopathology.61
Abdominal CT and ultrasonography studies may show renal AML, abdominal lymphadenopathy, lymphangioleiomyoma, ascites, and dilatation of the thoracic duct.18 AML occur predominantly in the kidney and liver and are recognized by their characteristic appearance consisting of areas of fatty density, intermixed with more dense areas and normal-appearing renal parenchyma (see Fig. 62-1C).18 Atypical AML lacking adipose tissue have a predominance of epithelioid LAM cells and radiologically may mimic renal cell carcinoma. Lymphangioleiomyomas appear as well-circumscribed masses of variable dimensions, comprising a wall and a central fluid-rich region (Fig. 62-1E).18 Diurnal variation in size of lymphangioleiomyomas has been demonstrated by CT and ultrasound, which may help differentiating them from malignant tumors and explain worsening of symptoms during day time.62,63
DIAGNOSIS
The characteristic CT scan appearance and its histologic features on open or thoracoscopic lung biopsy can diagnose LAM. Transbronchial lung biopsy may yield adequate sample size for pathologic diagnosis.64,65 The diagnosis of LAM should be strongly suspected in any woman who presents with progressive dyspnea, recurrent pneumothorax, or a chylous pleural effusion.1–5 The differential diagnosis includes pulmonary emphysema, asthma, chronic extrinsic allergic alveolitis, Langerhans cell histiocytosis, sarcoidosis, Birt–Hogg–Dubé syndrome, and follicular bronchiolitis. Definite LAM may be diagnosed in the presence of a characteristic HRCT and a lung biopsy showing the pathologic features of LAM or a characteristic lung HRCT and (1) angiomyolipoma, (2) chylous effusion, (3) lymphangioleiomyoma or lymphadenopathy, and (4) TSC.66 A diagnosis of probable LAM may be established in the presence of a characteristic HRCT and a compatible clinical history or a characteristic HRCT and angiomyolipoma or chylous effusions. Possible LAM may be diagnosed in the presence of a characteristic or compatible HRCT.66
Serum VEGF-D, a lymphangiogenic factor, is increased in the serum of patients with LAM compared to normal individuals and is a measure of lymphatic involvement in LAM.67–70 In the appropriate clinical and radiologic setting, a VEGF-D serum level equal or greater than 800 pg/mL is unlikely to be found in other cystic lung diseases and appears to be diagnostic of LAM.69,70
PROGNOSIS
The clinical course of LAM is highly variable. The estimated median transplant-free survival time for LAM patients in the United States is 29 years from symptom onset and 23 years from diagnosis.71 The estimated 10-year transplant-free survival is 86%. Age appears also to affect survival, as rapid decline in lung function is more common in younger premenopausal patients.49,72 Patients whose lung tissue shows predominance of cystic lesions tend to have worse lung function and prognosis than those with more LAM cell infiltrates.73
The severity of lung involvement in LAM may be assessed in patients who had a lung biopsy using the LAM Histology Score (LHS), which grades the extent of replacement of normal lung tissue by cystic lesions and LAM cell infiltrates.74 The amount of tissue involvement is graded semiquantitatively based on percent of lung tissue involved: LHS-1, <25%; LHS-2, 25% to 50%; and LHS-3, >50% lung tissue. LHS-2 and LHS-3 scores and the presence of hemosiderin-laden macrophages are associated with decreased survival.74 Patients with more cystic disease are likely to have lower FEV1 and DLCO, lower peak oxygen uptake (![]() O2max), and more exercise-induced hypoxemia.53,75
O2max), and more exercise-induced hypoxemia.53,75
The severity of lung disease in LAM can be also graded by HRCT. HRCT findings correlate with lung function tests, gas exchange, and exercise performance.53,56–59 HRCT computer analysis can quantify the percentage of lung volume affected by cysts and evaluate the texture of areas not involved with cysts.60,61 Using these methods, percentage of lung volume occupied by cysts was found to correlate with FEV1, residual volume, and DLCO.60,61
The simplest method of assessing the severity of lung disease in LAM is pulmonary function testing.1,49 Most patients have airflow obstruction and impaired gas exchange. Early in the disease, a significant number of patients may have normal spirometry or only mild airflow obstruction, along with a marked decrease in diffusion capacity. In these patients, the severity of disease is best graded by tests of gas exchange such as DLCO, arterial blood gases, A-a/O2 gradient, cardiopulmonary exercise testing, and 6-minute walk test.52,53,59 Exercise-induced hypoxemia may occur in the presence of near-normal DLCO and FEV1.53 Correlation between ![]() O2max and LHS scores, and between CT severity grade and A-a/O2 gradient, dead space/tidal volume ratio, and
O2max and LHS scores, and between CT severity grade and A-a/O2 gradient, dead space/tidal volume ratio, and ![]() O2max have been demonstrated.53,58–61 Rates of functional decline over time may help in defining the course of disease, that is, whether it is rapidly or slowly progressive. Sequential lung function testing every 3 to 6 months is warranted to assess the progression of disease.49,72 A positive response to bronchodilators occurs in 25% to 30% of LAM patients.75,76 Patients who respond to bronchodilators tend to have a predominantly cellular pattern of LAM lung lesions and greater rates of decline in FEV1.75 A low initial DLCO was also reported to be a predictor of accelerated loss of FEV1.77
O2max have been demonstrated.53,58–61 Rates of functional decline over time may help in defining the course of disease, that is, whether it is rapidly or slowly progressive. Sequential lung function testing every 3 to 6 months is warranted to assess the progression of disease.49,72 A positive response to bronchodilators occurs in 25% to 30% of LAM patients.75,76 Patients who respond to bronchodilators tend to have a predominantly cellular pattern of LAM lung lesions and greater rates of decline in FEV1.75 A low initial DLCO was also reported to be a predictor of accelerated loss of FEV1.77
OTHER PROGNOSTIC INDICATORS
There is some evidence that older age and/or menopause are associated with slower disease progression.49,72 Patients who present with exertional dyspnea and hemoptysis tend to have more severe disease, greater rates of progression of disease, and lower survival than those with a history of pneumothorax.78 This may be due to either a delay in diagnosis or an insidious course in those who present with dyspnea. Lymphatic involvement, for example, chylous effusions, lymphadenopathy, lymphangioleiomyomas, may be associated with a more severe form of disease.68,79,80 Correlation between LHS and the expression of VEGF-C has been reported.80 Serum levels of VEGF-D are especially elevated in patients with lymphatic abnormalities and show a correlation with DLCO and HRCT scan grading of severity of lung disease.68,79,80 Measurement of serum VEGF-D may be of value in establishing a diagnosis and grading the severity of disease and response to therapy.
TREATMENT
Treatment includes general principles of management and specific therapeutic interventions.
 GENERAL PRINCIPLES OF MANAGEMENT
GENERAL PRINCIPLES OF MANAGEMENT
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


