Fig. 28.1
Skull X-ray of a patient affected by PLCH. It shows two little osteolytic bone lesions easier to recognize in the lateral skull radiograph
Another important extrapulmonary complication is pulmonary hypertension. It is more severe in PLCH than in other interstitial lung diseases and histopathologically it is possible to recognize intimal fibrosis and remodeling of both venous and arterial systems [30]. Dauriat et al. estimated pulmonary hypertension in 92 % of 36 patients evaluated for lung transplantation [31]. Pulmonary hypertension is associated to poor prognosis so that it is necessary to screen all patients, especially those with excessive dyspnea and normal lung function tests, looking for ecocardiographic signs of pulmonary hypertension [30]. In selected cases cardiac catheterization would be necessary to confirm the diagnosis. When the diagnosis is made, therapy with vasodilators including phosphodiesterase inhibitors or endothelin receptor antagonists may be useful with an objective reduction in pulmonary artery pressure and improved exercise capacity. However, their potential capacity to worsen arterial oxygenation as a result of a greater imbalance in ventilation/perfusion must also be considered. This is possible because drugs can inhibit hypoxic pulmonary vasoconstriction. Le Pavec et al. reported their experience in a group of 29 PH-PLCH patients treated with the usual pulmonary hypertension therapies: endothelin receptor antagonist, phosphodiesterase 5 inhibitor and iloprost. Patients showed improvement in hemodynamics without oxygen worsening or pulmonary edema. However more studies are needed to evaluate safety and efficacy of usual pulmonary hypertension treatments in PH-PLCH [32]. Supplemental oxygen may be useful to correct hypoxemia while prostacyclin can cause severe pulmonary edema and should be used very cautiously in these patients because of the venous involvement.
Central nervous system (CNS) complications may occur in 1–11 % of LCH patients and can be subdivided clinically into two groups: the “neurodegenerative like” form which is characterized by neural cell loss and pyramidal syndrome; and the “mass lesion” form presenting as a space occupying lesion that may appear anywhere in the CNS. Very few studies are present in medical literature and an optimal treatment for CNS localization of the disease has not been defined. Tin et al. have described a good response to vinblastine, used in aggressive form of LCH, in patients with CNS mass lesion with a response rate of up to 70 %, while no effect has been described on neurodegenerative lesions [33].
Pulmonary Function Tests
Pulmonary function test findings are variable and the disease can be associated to a restrictive, obstructive or mixed pattern depending upon the course of the disease and prevalent anatomical lesions. In literature there are contrasting studies. According to Tazi et al. the obstructive pattern is the most frequent and there is evidence that flow–volume curve alterations are present in 50 % of patients, with the ratio of forced expiratory volume in 1 s (FEV1) to vital capacity (VC) diminished in 20–30 % of patients with recent onset of PLCH. This pattern may be related to the bronchial involvement characteristic of smokers, or to bronchiolar obstruction due to peribronchiolar fibrosis or inflammatory infiltrates [15]. On the contrary, Crausman et al. described a restrictive pattern in 11 patients of a cohort of 23 patients with early PLCH diagnosis. However, in advanced stages a restrictive pattern is usually the predominant pattern because of extensive lung fibrosis [21, 34]. At the time of diagnosis up to 20 % of patients may also have normal pulmonary function tests while, approximately 60–90 % of patients have low diffusing capacity for carbon monoxide (DLCO). Blood gas level at rest stay usually normal for a long time although it is possible to discover hypoxemia, and exercise limitation even in the earlier disease [15]. Canuet et al. tried to correlate lung function with HRCT lesions showing that the extent of cysts, is closely associated with the impairment of both lung function and gas exchange. A predominantly nodular pattern, suggestive of an active inflammatory disease, has instead only moderate functional consequences [35].
Tazi el al similarly described the correlation between lung function and HRCT lesions. They studied a group of 49 PLCH patients who experience a deterioration of lung function in 60 % of cases with a decline of FEV1 in 40 % of patients and a decline of DLCO in 50 % of the patients. The DLCO was reduced in some patients suggesting the hypothesis of pulmonary hypertension. However, according to other studies in literature the main pattern of lung function defect was represented by airway obstruction and this finding is consistent with the bronchiolar localization of pulmonary LCH lesions. The extension of cysts on HRCT scans correlated with a deterioration in lung function parameters so that serial lung function tests can be useful during follow-up, while routine serial HCRT seem to be less useful and expose these young patients to the dangers of radiation [36].
Chest Radiography
Most patients with PLCH exhibit chest radiographic abnormalities. In the earlier stages of the disease, it is common to find small nodules that typically range from 1 to 10 mm in diameter and have a bilateral and symmetric distribution on chest radiography. These nodules are characterized by irregular borders, and are present as single or merging nodules. The distribution of nodules is limited to upper/middle lung zones with sparing of the lung bases near the costo-phrenic sulci (Fig. 28.2). As the disease progresses, reticular-nodular abnormalities and cystic changes may predominate. As cysts become more numerous, nodules tend to occur less frequently [11]. End-stage PLCH is characterized by reticular areas of opacity that may progress to honeycomb lung and contiguous cystic cavities up to 2 cm diameter resulting radiographically indistinguishable from advanced emphysema or LAM. Especially in end-stage PLCH it is possible to find increased lung volumes on chest radiography and this can help to distinguish PLCH from other interstitial lung diseases which are instead characterized by reduced lung volumes (with LAM exception). Pneumothorax is known to be a complication of PLCH and may occur in the absence of other radiographic pulmonary abnormalities. Altogether, chest radiography has limited sensitivity and specificity for the detection and characterization of interstitial lung diseases, and in some cases of PLCH, chest X-ray may even appear normal. A rare finding in PLCH chest radiography is a solitary pulmonary nodule as described by Khoor et al. in a 45-year-old male cigarette smoker. Biopsy was performed to establish the nature of the nodule that showed the histologic and immunophenotypic characteristics of PLCH. Twenty-one years after its excision, appeared a new contralateral lung nodule which remained unchanged during 36 months of observation. Other rare finding in PLCH is represented by pulmonary artery prominence, due to pulmonary hypertension that may occasionally complicate PLCH [37].
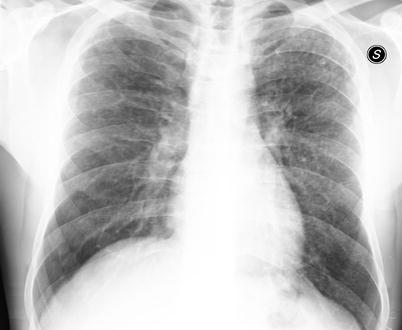
Fig. 28.2
Chest X-ray in a patient affected by PLCH. Small and poorly defined nodules are predominantely distributed in the upper and middle lung zones with the characteristic lower zones sparing
High Resolution Computed Tomography (HRCT)
HRCT is superior to radiography in demonstrating the morphology and distribution of lung abnormalities. Presentation patterns differ according to the stage of PLCH. In the early stages of the disease, as described by Brauner et al., a nodular pattern is usually recognizable, defined by the presence of multiple nodular opacities measuring 1–5 mm in diameter or larger [38]. Nodule sizes are usually less than 10 mm in diameter as also shown by Grenier et al. who observed nodules under 10 mm in diameter in the majority of CT scans from PLCH patients [39]. These small nodules, usually underestimated on chest X-rays, are characterized by irregular margins and are surrounded by normal lung parenchyma (Fig. 28.3). They may be profuse and are generally solid, although cavitation may occur with time. However, the predominant characteristic of lung nodules is their distribution. There is a topographic predominance in the upper and middle lung zones with relative sparing of the lung bases and most nodules show a centrilobular or peribronchial distribution, reflecting the bronchiole-centered development of PLCH lesions. According to Brauner et al. a temporary progression of these pulmonary nodules into cavitary nodules and then into cysts is possible [38]. In point of fact, follow-up scans showed a decreasing preponderance of nodules and an increasing number of thin-walled cysts. Cystic lesions tend to be initially small, with diameters of less than 10 mm, and thick-walled; then they become bigger with diameters up to 20 mm and thin-walled. Cyst formation progresses from cavitation within a centrilobular nodular lesion through an increasing bronchiolar dilatation resulting from granulomas destruction and supervening fibrosis on the edges of the lesion. Cyst distribution is also prevalent in upper lung zones where they can appear as round or ovoid cystic spaces or with bizarre shapes that may result from coalescence of adjacent cysts (Fig. 28.4a–c). Some of these cystic spaces may attain bulla sizes of up to 80 mm. Advanced disease is characterized by substantial architectural distortion due to cysts with few nodules [40] while late-stage disease is marked by the presence of large areas of honeycombing, predominantly in the upper lung zones. Some studies have described full or partial resolution of lesions occurring in patients with nodular lesions, indicating that nodules may be reversible, while cystic lesions remain unchanged or worsen with time [41]. There remains the question of whether a relationship exists between HRCT lesions and histopathological abnormalities. Soler et al. compared the nature of the main lesions on CT scans with that of lesions presented on biopsy samples in PLCH patients. They found that the usual, early-stage PLCH histopathological lesion consisted of florid granulomas that were mainly composed of typical LCs associated with macrophages and inflammatory cells, particularly lymphocytes and eosinophils. Interestingly, all patients showing a predominant nodular pattern on CT scans had florid granulomas in their lung tissue. In more advanced disease, cavitary granulomas were found in pulmonary samples, characterized by a prominent central cavity and few fibrotic changes, but still numerous LCs and inflammatory cells in their walls. In this case few cavitated nodules or thin-walled cysts were seen in CT scans. In late-stage PLCH fibrous cysts of variable size, demarcated by a fibrous ring of variable thickness, containing no LCs and few or no inflammatory cells are usually found [42]. Kim et al., studied a cohort of 27 PLCH biopsy proven patients, evaluating HRCT and histopathological findings at the time of surgical lung biopsy. The predominant CT pattern was represented by centrilobular nodules (10 patients) corresponding to peribronchiolar granulomas with LCs on biopsy. Nodules were typically present on upper lung zones with a random distribution. Thick, thin walled-cysts and bizarre shape cysts were respectively present in 4, 8 and 5 patients with a central cavity surrounded by a thin wall of LCs and eosinophils with a random distribution in the upper lung zones. Cystic lesions independently by the shape, can correspond to cavitated granulomas or to fibrous cysts. These two studies suggest that while it is possible to conclude that a nodular pattern on CT scans reflects a histopathologically active PLCH disease, no correlation is possible between histological and radiological findings in the case of patients with a cystic pattern because HRCT cannot differentiate between fibrous cysts and cavitary granulomas [41]. Canuet et al. tried to correlate lung function with HRCT lesions and showed that the distribution of cysts, was closely associated with an impairment of both lung function and gas exchange. A predominant nodular pattern, suggestive of an active inflammatory disease, has only moderate functional consequences and it was not significantly correlated with lung function parameters. A predominant cystic pattern was instead strongly correlated with lung function parameters [35]. Nodular changes may be present in several other lung diseases including sarcoidosis, silicosis, tuberculosis, RB-ILD, hypersensitivity pneumonitis or metastatic disease [42]. In some of these cases the radiologic differential diagnosis with PLCH may turn into a dilemma. All these conditions are in fact characterized by a centrilobular nodular pattern of presentation on CT scans. However, the nodule distribution is one aid to a correct diagnosis. Perilymphatic distribution is typical of sarcoidosis in which nodules are predominantly present in interlobular septa, peribronchovascular and sub-pleuric spaces. Random distribution of nodules is possible in miliary tuberculosis and hematogenous metastases and it is characterized by a random location of the nodules in the secondary pulmonary lobule. In some cases it can be difficult to differentiate metastatic nodules from PLCH nodules although metastatic nodules are often surrounded by a halo of ground glass attenuation useful to make the differential diagnosis. In other cases only lung biopsy may have a discriminative importance [43]. When only cysts are seen on the CT scan, a differential diagnosis has to be made between PLCH and other lung diseases with a cystic CT-scan pattern such as idiophatic pulmonary fibrosis (IPF), emphysema, bronchiectasis and lymphangioleiomyomatosis (LAM). Differential diagnosis between IPF and PLCH is necessary when CT scans indicate honeycombing, defined as the presence of air-filled cystic spaces that often predominate in a peripheral sub-pleural location. These cysts can be of different sizes with a wall thickness between 1 and 3 mm, consisting of fibrous tissue lined by bronchiolar epithelium, which is shared by adjacent cysts. This is specific for honeycombing and not seen in cysts that are found in PLCH. However, a differential diagnosis can be easier because honeycombing is often associated with other findings of pulmonary fibrosis such as reticular opacities, irregular sub-pleural and peribronchovascular thickening and traction bronchiectasis while PLCH cysts are surrounded by normal lung zones. The most important difference between the two entities remains the lower zones sparing, typical of PLCH, while IPF is characterized by bi-basal and sub-pleural distribution of the cysts with the typical apical-basis gradient. Pulmonary emphysema is defined as a permanent, abnormal enlargement of airspaces distal to the terminal bronchiole accompanied by the destruction of the alveolar walls. It is usually easy to differentiate emphysema by the focal areas of low attenuation that are found in PLCH, contrasted by surrounding normal lung, and by the absence of walls [44]. Bronchiectasies are localized, irreversible bronchial dilatation, very often with thickening of the bronchial wall that may be mistaken for cystic airspace disease when a dilated airway is viewed “frontally”. It can be differentiated from cystic lung disease by the presence of an adjacent blood vessel suggesting a bronchovascular unit rather than a cystic air space [45]. LAM is characterized on CT scans by the presence of thin-walled cysts of variable size from 2 to 40 mm diameter. Vessels can be seen at the periphery of the cysts, unlike emphysema where vessels may be found in the center of the lesion. The most important difference between LAM and PLCH lies in the distribution of cysts: with LAM cysts involving uniformly all regions of the lungs without sparing the costo-phrenic angles [45, 46].
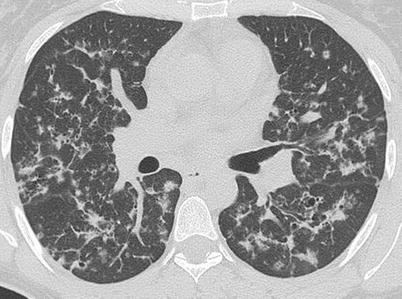
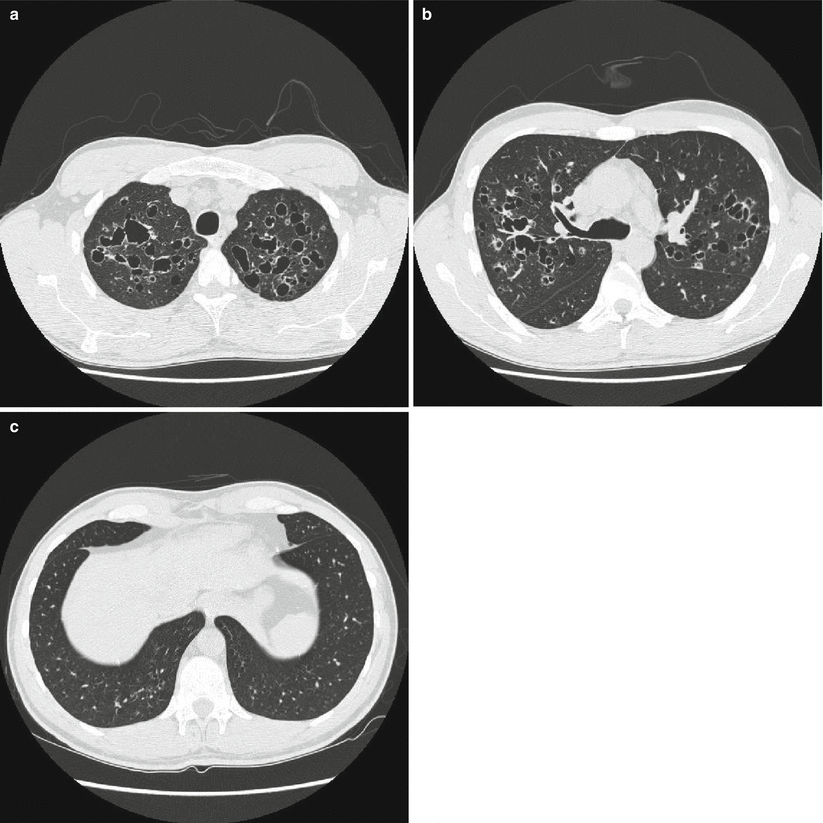
Fig. 28.3
HRCT of the lungs of a patient affected by PLCH, showing a predominant nodular pattern. It is possible to describe centrilobular and peribronchiolar nodules, some of which are cavitated (Courtesy of Professor N. Sverzellati. University of Parma, Italy)
Fig. 28.4
HRCT of the lungs of a patient affected by PLCH showing a predominant cystic pattern. The cysts are characterized by variable wall thickness and bizarre shapes (a). It is possible to identify centrilobular and bronchiolocentric nodules, some of which start to cavitate (b). Note the predominant upper lung involvement with relative sparing of the lung bases (c)
Bronchoscopy and Bronchoalveolar Lavage (BAL)
Bronchoscopy is a well tolerated procedure that can provide further information for the correct diagnosis of PLCH. In PLCH the bronchial tree is usually either normal on gross examination or there are signs of non-specific inflammation due to smoking. It is possible to identify different nonspecific features in the bronchoalveolar lavage fluid of PLCH patients, such as an increased number of cells with a marked predominance of alveolar macrophages, a decreased CD4/CD8 ratio or increased levels of eosinophil cells. These features are typically present in the BAL fluid of smokers where there is no evidence of interstitial lung disease, so it is necessary to look for other disease markers in the BAL fluid. LCs, identified by staining with antibodies against CD1a and S-100 antigens on the cell surface have been proposed as PLCH markers. Casolaro et al. have demonstrated the presence of LCs in the bronchoalveolar lavage of smokers without interstitial lung diseases, concluding that cigarette smoking is associated with an expansion in the population of LCs on the epithelial surface of the lower respiratory tract [47]. In addition, Tazikawa et al. have described the presence of LCs in patients affected by idiopathic pulmonary fibrosis, sarcoidosis and other fibrotic lung disorders so it would seem that LCs in bronchoalveolar lavage are not so specific for diagnosing PLCH [48]. However, Tazi et al. demonstrated that even if an increased level of LCs may be present in other pulmonary diseases it is possible to fix a threshold of 5 % LCs to define the test as specific, although the sensitivity remains quite low (<25 %) [15]. Smetana et al. demonstrated the presence of another cell marker in the BAL fluid of PLCH patients named Langerin (CD207). This is a protein specific for LCs present in the skin and in the epithelia of small airways and bronchioles. They compared the percentage of CD1a and Langerin positive cells in PLCH with other fibrotic lung disorders such as sarcoidosis and IPF. This analysis showed that the numbers of CD1a and Langerin positive cells were almost identical in all the tested cases, underlying the potential relevance of Langerin in improving the usefulness of BAL in the diagnosis of PLCH [49], although its real diagnostic utility has yet to be assessed in clinical studies. Bronchoalveolar lavage rarely establishes a definitive diagnosis of PLCH in adults, despite this, it is helpful in the differential diagnosis of infectious diseases characterized by the presence of excavated nodules, such as Pneumocystis jiroveci pneumonia, or in patients without a typical radiological pattern where it can orientate towards the diagnosis to PLCH showing high alveolar macrophage counts or increased levels of LCs.
Lung Biopsy
Transbronchial lung biopsy has a limited role in the diagnostic workup for PLCH as confirmed by Vassallo et al. in a study of 102 patients with histological diagnoses of PLCH. Among 29 patients who underwent transbronchial lung biopsy the findings were diagnostic only in six patients [50]. However, transbronchial lung biopsy remains useful for differential diagnosis by excluding other disorders, such as sarcoidosis. Even if HRCT is suggestive of PLCH in the great majority of patients with typical clinical manifestations, some cases are difficult to interpret. In patients with systemic symptoms and cavitated pulmonary nodules, patients with suspected pulmonary metastases or in female patients in differential diagnosis with LAM, a definitive diagnosis relies on pulmonary biopsy. In those cases where diagnosis is uncertain, surgical biopsy may permit a diagnosis of PLCH demonstrating the presence of the characteristic lesions. However, lung biopsy should not be performed in patients with extensive destructive lesions given the increased surgical risk [15]. In a patient with suspected PLCH and an extra-pulmonary lesion with compatible HRCT findings, a diagnosis may be provided by a biopsy of this lesion. However, the majority of adults have isolated PLCH, which makes a surgical lung biopsy using video-assisted thoracoscopy or open thoracotomy useful for a correct diagnosis. Since the lesions are focal the specimens should be sufficiently large to take an adequate quantity of material, preferably from different areas of the lung. Lung biopsy can generally be avoided when HRCT findings are characteristic and concordant with the clinical history [42].
Pathology
Gross lung tissue specimens in PLCH may exhibit different features according to the stage of the disease at the time of biopsy. In the earlier stages nodules are seen as focal lesions with irregular and stellate borders. In advanced disease phase, the predominant finding is a hyperinflated lung with cysts and honeycombing formation [42]. On microscopic examination, the characteristic early lesions in PLCH are situated in terminal and respiratory bronchioles and are formed by activated LCs organized into a loose granuloma associated with lymphocytes and inflammatory cells like eosinophils and macrophages (Fig. 28.5a, b) [10]. The morphology of LCs found in these inflammatory nodular lesions is generally similar to that of LCs in normal tissues: they are of medium size with elongated nuclei, display multiple cytoplasmic extensions and pale cytoplasm which contains few phagocytic vacuoles. A definitive identification of LCs in these inflammatory lesions is possible by immunohistochemical staining with monoclonal antibodies directed against the membrane antigen CD1 (Fig. 28.6) or by the recognition of Birbeck granules, identifiable through electron microscopic visualization. Birbeck granules are intracytoplasmic organells that may be involved in the intracytoplasmic transport of antigens captured by LCs. The histology of PLCH granulomas varies according to the particular stage of the disease even if lesions of different ages can be found in the same lung biopsy specimen. The lesions are focal, poorly demarcated, separated by apparently normal lung parenchyma, and centered on the terminal and respiratory bronchioles destroying the airway walls, thus giving the impression that PLCH mimics bronchiolitis rather than being a diffuse infiltrating lung disease. At this stage, LCs form a compact central granuloma surrounded by variable numbers of lymphocytes, eosinophils and macrophages, which extend to adjacent alveolar structures. This lesion may evolve forming a cavity that results from the residual lumen of the bronchiole destroyed by granulomatous reaction and not from its necrosis. The PLCH granulomas are poorly demarcated and extend in adjacent alveolar structures that often contain pigmented macrophages, producing RB-ILD-like changes or a desquamative interstitial pneumonia-like pattern. In lesions of intermediate age, there are few LCs while lymphocytes, macrophages and neutrophils are still present in LCH granuloma. In late-stage lesions LCs are almost absent and there are more macrophages containing pigment or lipid inclusions [10]. The lesions are then replaced by stellar fibrotic scars or by confluent adjacent cysts giving the characteristic aspect (Fig. 28.4). Interestingly, in uninvolved areas, the lung structure seems to be normal or characterized by common smoking-related abnormalities, such as respiratory bronchiolitis and increased levels of pigmented macrophages infiltrating the bronchiole walls [15].
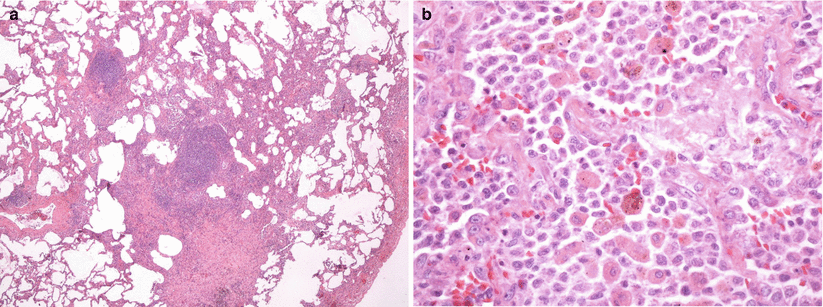
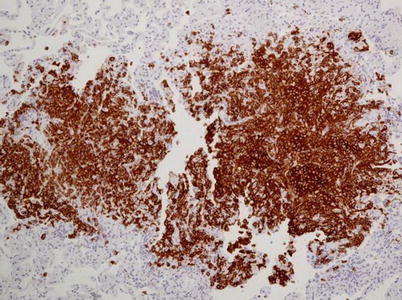
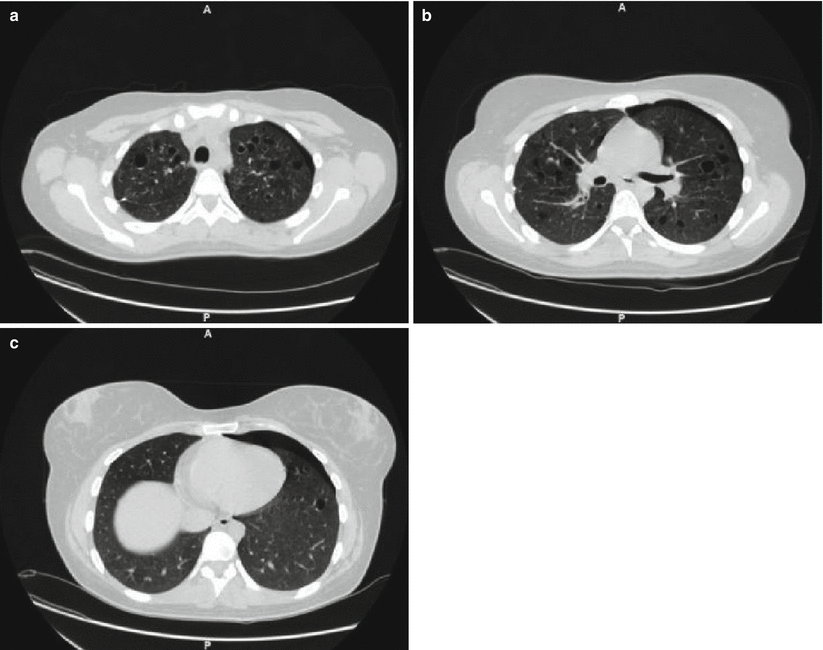
Fig. 28.5
LCH with centrolobular nodules with irregular margins (a) and aggregates of Langerhans cells together with “golden” macrophages and eosinophils (b) (Courtesy of Professor G. Rossi. University of Modena-Reggio Emilia, Italy)
Fig. 28.6
CD1a further highlights Langerhans cells at immunohistochemistry (Courtesy of Professor G. Rossi. University of Modena-Reggio Emilia, Italy)
Fig. 28.7
(a) High-resolution CT scan demonstrates a small left pneumothorax and bilateral irregular cysts, with thin walls. (b) Some of these cysts appear to coalesce into larger and irregular structures with bizarre shapes. (c) Typical lesion distribution with relative sparing of lower lobes
Diagnosis
When PLCH is suspected on the basis of clinical-radiological and anamnestic features it is necessary to continue searching for other organ involvement of the disease. Skeletal X rays are necessary to investigate the presence of bone lesions, serological investigations to evidence increased plasma osmolarity and RM of the brain can be helpful to identify possible involvement of hypothalamic region.
Treatment Options
The recruitment of a sufficient number of patients for controlled therapeutic trials has been hampered by the low incidence of PLCH and its clinical stability. To date, no randomized trials of therapy for adult PLCH have been reported. All data regarding the effectiveness of PLCH treatment are derived from observational studies, case reports and expert opinions. The association between PLCH and smoking suggests that cigarette smoke plays a role in the pathogenesis of the disease. Therefore, it is necessary that patients stop smoking and this should be encouraged, especially in heavy smokers, through smoking cessation programs [15]. Some case reports have shown that this alone can lead to an improvement in the clinical and radiographic findings or even in the resolution of the disease [50]. Mogulkoc et al. described two cases of PLCH in smokers characterized by the presence of nodules some of which were cavitated or formed small cysts on CT scans and by a reduced DLCO. After smoking cessation there was an objective radiological improvement with reduction of nodules and a functional improvement with DLCO increase [20]. Similarly, Negrin-Dastis [51] described a PLCH case with total regression of radiological lesions after 12 years of smoking cessation. Because of the rare incidence of PLCH and the unpredictable course of the disease, there are no reliable data regarding the efficacy of smoking cessation on disease resolution as showed by Tazi et al. [52] who reported four PLCH cases with biopsy proven PLCH diagnosis: all of them were smokers and their disease initially underwent regression just by smoking cessation and subsequently developed reactivation with the appearance of new nodules on CT scans, thus requiring corticosteroid therapy. However, other studies provide contrasting data, describing cases where the disease has worsened despite smoking cessation [52, 53] and recently Tazi et al. have described how smoking cessation did not modify the pulmonary LCH outcomes in a group of 49 LCH adult patients [36]. This leads us to conclude that smoking cessation might lead to an improvement in the clinical and radiographic findings or even to the resolution of the disease but it should also be remembered that the disease can improve by virtue of its natural course and there is, as yet, no definitive proof that smoking cessation affects the outcome of the disease. Nonetheless, smoking cessation is considered as a first step in the treatment of PLCH. Failure to prevent the progression of the disease by this means is generally followed by steroid treatment. The rationale of using corticosteroids, especially in the early stages of the disease in which nodular lesions are the predominant features, is based on the possibility of accelerating the resolution of the associated granulomatous and inflammatory processes while in advanced stages the presence of fibrosis may explain a lack of response to therapy. On this basis, it has been suggested that initiating corticosteroid therapy in symptomatic PLCH with a predominant nodular pattern on HRCT scans can improve radiological and clinical features. Usually prednisone or prednisolone are administered at a starting dose of 0.5–1 mg/kg/day tapered over 6–12 months [15]. In a group of 42 PLCH patients treated with corticosteroids, Schonfeld and coworkers demonstrated clinical and radiographic improvements, although they did not observe any significant changes in respiratory function [54]. In case of disease progression in spite of a 6-month period of steroid treatment, the disease can be treated with chemotherapy. The agents used include vinblastine, mercaptopurine, cyclophosphamide or, more recently, cladribine (2-chlorodeoxyadenosine).
In the 1960s, chemotherapy was used to treat LCH in children because it was thought to be a malignant process. Single agents such as methotrexate, 6-mercaptopurine, vinblastine and vincristine were initially and successfully used in pediatric patients and these encouraging results made possible to start new trials. Different perspective randomized trials were conducted by the Histiocyte Society: in the first study the efficacy of vinblastine or etoposide in combination with prednisolone was compared. For 24 weeks, patients assumed vinblastine (6 mg/m2) intravenously every week, or etoposide (150 mg/m2/day) intravenously for 3 days every 3 weeks and a single initial dose of corticosteroids. There was no difference in survival or disease reactivation rates but the absence of response after 6 weeks of treatment was related to poor prognosis with increased mortality. The second trial conducted by the Histiocyte Society was carried out on 193 randomized LCH children divided into two groups, the first group receiving vinblastine, prednisolone and mercaptopurine, the second receiving the same therapy with the addition of etoposide. The dosage was as follows: first group, initial treatment of continuous oral prednisone (40 mg/m2 daily in three doses for 4 weeks tapering over 2 weeks) and vinblastine (6 mg/m2 intravenous bolus weekly for 6 weeks); while the second group received the same therapy with the addition of etoposide (150 mg/m2 per day, 1-h infusion weekly for 6 weeks). At the 6th week, the continuation therapy was 6-mercaptopurine (50 mg/m2 daily orally) and pulses of oral prednisone (40 mg/m2 daily in three doses, days 1–5) and vinblastine (6 mg/m2 per day once every 3 weeks) in the first group, while the second group added vinblastine every 3 weeks. The total duration of treatment was 24 weeks. This trial showed that more intensive treatment increases response rates and reduces mortality from LCH [55]. The Histiocyte Society has recently closed its third clinical trial conduced with the aim of assessing whether the addition of methotrexate to prednisolone and vinblastine and increasing treatment duration to 12 months could reduce relapse rates. Even though all of these studies were performed on a pediatric population, they suggest that these agents may have a role in the treatment of LCH in adults with pulmonary and/or multisystemic involvement considering PLCH as a single system disease with a “risk organ” involvement [56]. In multisystem LCH, often refractory to treatment and characterized by frequent relapse, there is no current standard salvage regimen. Recently, however, cladribine has been used as a second-line treatment for both children and adults with good response. Cladribine is a purine nucleoside analogue with selective toxicity to lymphocytes and monocytes, which acts by interfering with single-stranded DNA repair and synthesis in lymphocytes and monocytes. Aerni et al. described a case of LCH with pulmonary involvement that responded well to cladribine treatment, suggesting the possibility of its use in selected cases [57]. Other therapies have been proposed for LCHs, including oral acitretin, which is a Vitamin A analogue. Derenzini et al. treated a group of seven patients, three suffering from multisystem and four from single-system LCH, with MACOP-B. This chemotherapy regimen is used for non-Hodgkin’s lymphoma and consists of a combination of prednisolone, vincristine, bleomycin, metotrexate, doxorubicin and ciclophosphamide. A 100 % response rate in all seven adult patients was reported [58].
However PLCH treatment is not standardized, and data regarding the effectiveness of treatment are derived from observational studies, case reports and expert opinion. More studies are needed regarding less toxic and more effective treatments.
Another important treatment to be considered is pleurodesis in cases of recurrent pneumothorax following the rupture of a cystic lesion. Mendez et al., demonstrated the superiority of pleurodesis to tube thoracostomy alone in ipsilateral recurrence of pneumothorax [59].
Lung transplantation is performed in selected patients with progressive disease that is refractory to other forms of treatment, including patients with severe pulmonary hypertension unresponsive to vasodilator therapy, and when severe respiratory failure develops. Etienne et al. performed lung transplantation in seven adult LCH and observed resolution in five of these patients while the other two patients have suffered recurrence of LCH in the grafted lung. These two patients resumed smoking after transplantation and had extrapulmonary localization of the disease [60]. A retrospective multicenter study on 39 patients who underwent lung transplantation for end-stage PLCH described a recurrence rate in the allograft as high as 20.5 %. The presence of the extrapulmonary disease before transplantation and a resumption of smoking have been described as risk factors for recurrent disease so that, although lung transplantation may be an efficient treatment for end stage LCH, the risk of recurrence must be considered [31].
Course and Prognosis
PLCH is characterized by an unpredictable course in the individual patient ranging from an asymptomatic and stable course to a progressive debilitating disease that leads to respiratory failure and death over a period of months. Identification of patients with poor prognosis could be useful in deciding who will benefit from aggressive treatment early in the course of disease. According to Delobbe et al., older age, lower FEV1/FVC ratio at diagnosis and prolonged corticosteroid therapy suggest an adverse prognosis [61]. However, in this study the diagnosis of PLCH was not confirmed by lung biopsy and some patients included were children. Other studies in literature suggest that PLCH patients are at increased risk of developing bronchogenic carcinoma and hematological malignancies although such occurrences may be merely coincidental [62]. In a more recent study Vassallo et al. studied a cohort of 102 PLCH patients and the median survival was estimated to be of 12.5 years showing that adult patients affected by PLCH have a shorter survival than general population. Reduced DLCO or severe COPD due to concomitant cigarette smoke were considered as possible negative prognostic factors [50]. Pulmonary hypertension is an unrecognized complication of PLCH that is related to poor prognosis [63]. Thus, it is important to estimate pulmonary hypertension by echocardiography at the time of diagnosis and afterwards in follow up controls. When pulmonary hypertension is suspected on the basis of echocardiography, and especially when the estimated PAP is higher than 40 mmHg, a cardiac catheterization it would be useful to confirm and define the severity of pulmonary hypertension [21]. Multiorgan involvement may be characterized by poor prognosis and for correct management of PLCH is necessary to investigate other possible organ involvement. The diagnostic approach to these patients should include skeletal X-rays to show possible bone disease and gadolinium enhanced magnetic resonance imaging of the brain to identify potential involvement of the hypothalamic region. In recent years, fluorodeoxyglucose (FDG) PET scan imaging has been proposed to differentiate malignant pulmonary nodular lesions from benign ones. However, false positive FDG-PET scan have been demonstrated in other conditions such as active infections, noninfectious inflammatory processes, benign neoplasm and interstitial lung diseases such as sarcoidosis. In a cohort of 11 patients with PLCH diagnosis, PET-scan-positive patients had predominantly nodular lung disease, while PET scan negative patients had mainly a cystic lung disease. However, it was not possible to distinguish between the benign inflammatory nodular lesions of PLCH and malignant lesions because the pulmonary nodules and some cystic lesions, frequently demonstrate standardized uptake value (SUV >2.5) similar to malignant lesions [64]. Phillips et al. compared both the capacity of different imaging techniques to determinate the extent of LCH and the effectiveness of therapy. A decreased FDG uptake following therapy suggested a role for FDG-PET in detecting disease activity and early response to therapy with greater accuracy than other imaging modalities in patients with LCH affecting bones and soft tissues [65]. However, it is necessary to have more perspective data to guide the clinical use of FDG-PET in the diagnosis and follow-up of LCH. After PLCH diagnosis is made, it is necessary to follow the course of the disease evaluating clinical parameters, chest radiography or better HRCT, and pulmonary function, initially at intervals of no more than 6 months. HRCT scanning has proven to be useful in understanding the evolution of the pathological lesions confirmed by the study of Soler et al. in which a correlation between the extent of nodular abnormalities and the density of florid granulomatous lesions in lung tissue was demonstrated. Long term follow up of PLCH patients is recommended because even after years of apparent quiescence, lung function can deteriorate and new nodular lesions can occur, suggesting a reactivation of the disease [42]. Case reports of PLCH in pregnant women have been reported. Pregnancy does not seem to influence the course of the disease, except for the appearance of exacerbations of LCH-related diabetes insipidus. Pregnancy is not contraindicated in PLCH women unless there is a severe respiratory failure [10].
RB-ILD
Introduction
Bronchiolitis is a generic term used to describe an inflammatory process regarding the small airways that may be the consequence of cigarette smoke, infections, aspiration, environmental agents, drugs or underlying systemic disorders such as connective tissue diseases and transplantation rejection [66]. In 1974, Niewoehner described the presence of inflammatory changes in the peripheral airways of a group of young smokers who had died of sudden death. The post-mortem findings showed the presence of clusters of pigmented macrophages in respiratory bronchioles and neighboring alveoli [67]. These changes are common in cigarette smokers and the term respiratory bronchiolitis (RB) is appropriately used to indicate small airways inflammation in smokers. In 1987 Myers et al., studied six patients with clinical, functional and radiological features suggestive of interstitial lung disease who underwent lung biopsy. The major pathologic findings were the presence of respiratory bronchiolitis, characterized by clusters of pigmented alveolar macrophages within respiratory bronchioles, and, in addition, a mild chronic interstitial inflammatory infiltrate of bronchiolar walls associated with hyperplasia of alveolar epithelial cells [68]. These were the first cases, described in literature, of respiratory bronchiolitis associated with an inflammatory involvement of the interstitium. The term “respiratory bronchiolitis with associated interstitial lung disease” (RB-ILD) appeared in 1989 in a study of Yousem et al. concerning the histopathological differences between this new entity and other interstitial lung diseases. This and other studies, looking at the histopathological differences between RB and RB-ILD, have concluded that RB-ILD is usually associated with a greater extension of fibrosis, even though lungs of smokers affected by RB, may also show mild alveolar fibrosis. The result is that differential diagnosis between RB and RB-ILD, exclusively based on histopathological and/or radiological findings, can be very difficult [69]. Thus, the diagnostic suspect should also be based on clinical presentation. RB is usually asymptomatic while symptoms, such as dry cough and dyspnea, are more often present in RB-ILD [67].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


