Pulmonary Langerhans’ Cell Histiocytosis
INTRODUCTION
Pulmonary Langerhans’ cell histiocytosis is part of a large spectrum of disorders characterized by abnormal organ infiltration by Langerhans’ cells, which are highly differentiated cells in the monocyte-macrophage line that are also found in the dermis of the skin, reticuloendothelial system, pleura, and lung. Clinically, these disorders vary greatly, ranging from mild, single-organ disease to acute, disseminated life-threatening presentations. Depending on the sites involved and severity, the entity now referred to as Langerhans’ cell histiocytosis has been previously defined as eosinophilic granuloma, Hand–Schüller–Christian disease, and Letterer–Siwe disease. A more recent and simplified system of classification includes Langerhans’ cell histiocytosis with single-organ involvement or with multisystem involvement.1
Pulmonary Langerhans’ cell histiocytosis is an uncommon, smoking-related, interstitial lung disease that primarily affects young adults. Usually, lung involvement occurs in isolation; less frequently, involvement of other systems, for example, bone, skin, pituitary gland, is seen. Although there is some similarity to other diffuse interstitial lung diseases, pulmonary Langerhans’ cell histiocytosis, as a specific disease entity, is distinct in its clinical, radiologic, and pathologic manifestations.
EPIDEMIOLOGY
The true incidence and prevalence of pulmonary Langerhans’ cell histiocytosis are unknown. Studies in which diagnoses were confirmed by lung biopsy showed that pulmonary Langerhans’ cell histiocytosis is an uncommon, if not rare, disease.2,3 A Japanese study of discharge diagnoses in hospitals with 200 beds estimated the disease prevalence at 0.27 and 0.07 per 100,000 population in males and females, respectively.4 These reports may underestimate the true incidence of the disease, as lung biopsy is not performed in all cases of pulmonary Langerhans’ cell histiocytosis, and some patients exhibit no symptoms or experience spontaneous remission. Occupational or geographical predisposition has not been reported. Of note, nearly all affected persons report a current or prior smoking history. Thus, tobacco smoke is thought to play a key role in the pathogenesis of pulmonary Langerhans’ cell histiocytosis of adulthood. Other diffuse parenchymal lung diseases associated with cigarette smoking are respiratory bronchiolitis-associated interstitial lung disease and desquamative interstitial pneumonitis.5
Most patients with pulmonary Langerhans’ cell histiocytosis present to medical attention in young adulthood (20–40 years of age). Pulmonary Langerhans’ cell histiocytosis, however, may present in any age group. Older literature suggested a male preponderance; however, recent literature suggests an equal sex distribution, with increasing presentations in middle age. In general, women tend to present at an older age than men. These differences in prevalence may reflect the changing smoking habits of women in our society. For unknown reasons, whites are affected much more commonly than blacks or Asians, in whom this disease is very rare.
Pulmonary Langerhans’ cell histiocytosis has reportedly been associated with a number of malignancies and may be a premalignant condition. Lymphoma, both Hodgkin’s and non-Hodgkin’s, and other hematologic and solid cancers have been reported in association with pulmonary Langerhans’ cell histiocytosis.6 However, the evidence regarding this association is inconclusive. Malignancies may precede, follow, or occur concomitantly with the diagnosis of the interstitial lung disease. The carcinogenic effects of cigarette smoke are probably responsible for some of these tumors; thus, other components of tobacco may be responsible for pulmonary Langerhans’ cell histiocytosis. Given the complexity of factors involved in the pathogenesis of cancer and pulmonary Langerhans’ cell histiocytosis, it has been difficult to define the effects of tobacco on the pathogenesis of malignancies in patients with pulmonary Langerhans’ cell histiocytosis. Possibly, shared genetic predisposition factors may also have a role in the development of malignancies in patients with the disorder.7
NATURAL HISTORY AND CLINICAL PRESENTATION
Patients with pulmonary Langerhans’ cell histiocytosis come to medical attention in a variety of ways: as an incidental diagnosis that is suggested by a screening chest radiograph, after pneumothorax, or with respiratory or constitutional symptoms. Symptomatic patients most often have a nonproductive cough (56%–70%), dyspnea (40%), chest pain (10%–21%), fatigue (~30%), weight loss (20%–30%), and fever (15%). Pleuritic pain and acute dyspnea with a spontaneous pneumothorax can be a recurrent problem in as many as 25% of patients. Pleural thickening or effusion is rarely seen in the absence of a history of pneumothorax. Hemoptysis (13%) is occasionally reported, and it should prompt consideration of superimposed infection (e.g., Aspergillus) or tumor.8
Cystic bone lesions are present in 4% to 20% of patients with pulmonary Langerhans’ cell histiocytosis and may produce localized pain or a pathologic bone fracture. The precise number of patients with bone lesions is not known because complete bone surveys are not routinely performed. Skeletal involvement may be either the sole symptomatic manifestation of pulmonary Langerhans’ cell histiocytosis or may precede the more typical pulmonary manifestations. The radiographic pattern is not diagnostic. In most instances, the lesions are solitary and affect the flat bones. Central nervous system involvement with diabetes insipidus (approximately 15% of patients) is also seen and is believed to portend a poor prognosis. Skin involvement may also be present in adults with pulmonary Langerhans’ cell histiocytosis.
Skin lesions are usually erythematous, maculopapular, or nodular. In these patients, the scalp is often involved by characteristic seborrheic and crusted lesions.9
The physical examination is usually unremarkable. On chest examination, crackles are uncommon. Digital clubbing is also uncommon. Secondary pulmonary hypertension (PH) may occur and is probably underrecognized. Manifestations of cor pulmonale are seen in advanced stages. Routine laboratory studies are usually unrevealing; the peripheral eosinophil count is normal.1
PATHOGENESIS
The pathogenesis of adult pulmonary Langerhans’ cell histiocytosis is still poorly understood. However, the nearly universal association with cigarette smoking strongly implies causation. Smoke may activate alveolar macrophages through bombesin-like peptides. Bombesin is a neuropeptide produced by neuroendocrine cells, which are increased in the lungs of smokers.10 Bombesin-like peptides are chemotactic for monocytes, are mitogenic for epithelial cells and fibroblasts, and stimulate cytokine secretion. Several antigens in cigarette smoke, including tobacco glycoprotein, may stimulate macrophage and epithelial cell production of cytokines, such as granulocyte-macrophage colony-stimulating factor (GM-CSF), dendritic cell chemokines, for example, chemokine (C-C motif) ligand 20 (CCL20), osteopontin, and tumor necrosis factor-α (TNF-α) that promote recruitment and activation of Langerhans’ cells.11 In fact, GM-CSF and TNF-α have been found in the lesions of patients with Langerhans’ cell histiocytosis and have been shown to facilitate the in vitro generation of Langerhans’ cells from CD34+ hematopoietic stem cells.12 TNF-α and other cytokines, for example, tumor necrosis factor-β (TNF-β), may also stimulate fibroblasts leading to fibrosis.13 Moreover, tobacco glycoprotein may also cause an abnormal differentiation of T lymphocytes and a reduction in interleukin (IL)-2 release by lymphocytes, thereby enhancing survival or proliferation of Langerhans’ cells (Fig. 61-1).14
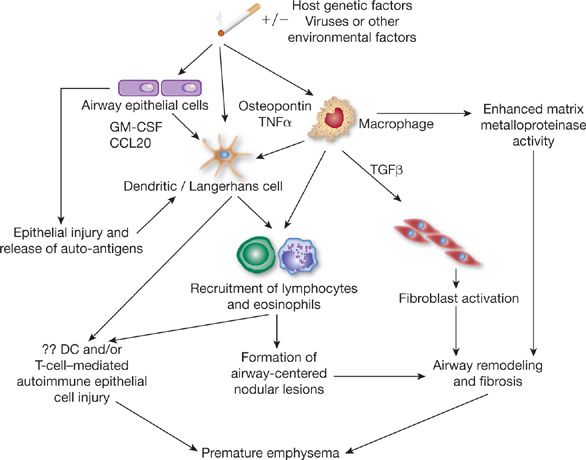
Figure 61-1 The primary event in the pathogenesis of pulmonary Langerhans’ cell histiocytosis probably involves cigarette-smoke–induced recruitment to the lung and activation of Langerhans’ cells, a process that may result from a variety of potential mechanisms. Antigens in cigarette smoke, including tobacco glycoprotein (TGP), may stimulate alveolar macrophages and epithelial cells to produce cytokines or other factors that enhance recruitment and activation of Langerhans’ cells. Cigarette smoke may also directly activate Langerhans’ cells to secrete cytokines (such as TNF or GM-CSF) that mediate local accumulation of inflammatory cells, with resultant formation of nodules. Uptake of cigarette-smoke antigens by alveolar macrophages or Langerhans’ cells may also promote local expansion of T lymphocytes and further inflammation. Through the action of tobacco glycoprotein, reduced interleukin-2 secretion by lymphocytes may occur, thereby enhancing local survival and proliferation of Langerhans’ cells. T lymphocytes may further stimulate B-lymphocyte activation, promoting secretion of antibodies and immune-complex formation. Fibroblast activation and fibrosis may result from the local synthesis of tumor growth factor-β (TGF-β) and by alveolar macrophages. (Reproduced with permission from Suri HS, Yi ES, Nowakowski GS, Vassallo R. Pulmonary langerhans cell histiocytosis. Orphanet J Rare Dis. 2012;7:16.)
Abnormalities in immune function, with a nonspecific increase in immunoglobulin G (IgG) in bronchoalveolar fluid, circulating and tissue-bound immune complexes, and abnormalities in T-cell function, have been observed in association with pulmonary Langerhans’ cell histiocytosis and may be important in the pathophysiology of this disorder.15 It is possible, however, that these findings represent nonspecific consequences of a generalized activation of immune effector cells.
Recent studies report high serum levels of IL-17A during active Langerhans’ cell histiocytosis, IL-17A synthesis by dendritic cells of patients with multisystem Langerhans’ cell histiocytosis, and an IL-17–dependent pathway for dendritic cells fusion, showing that IL-17 may play a role in pathogenesis, although its significance in pulmonary Langerhans’ cell histiocytosis is not known.16 Some studies suggest that the pathogenesis of pulmonary Langerhans’ cell histiocytosis entails alterations of the expression of the adhesion molecules that regulate interactions between white blood cells and endothelial cells.17,18 One important adhesion molecule for neutrophils that is expressed by endothelial cells is intercellular adhesion molecule-1 (ICAM-1). ICAM-1 expression by Langerhans’ cells has been demonstrated in biopsy specimens of subjects with Langerhans’ cell histiocytosis. Expression of other leukocyte adhesion molecules, such as the β1 and β2 integrins, has also been noted.19 The significance of these findings and their relevance to pulmonary Langerhans’ cell histiocytosis remain to be elucidated.
Alternatively, a viral infection has been suggested as the underlying cause of generalized Langerhans’ cell histiocytosis. However, there are no convincing data to suggest a role for viral infection as a cause of pulmonary Langerhans’ cell histiocytosis.20 Although clonality of histiocytes has been shown in children and adults with multisystem Langerhans’ cell histiocytosis or unifocal bone disease, pulmonary Langerhans’ cell histiocytosis appears to be primarily a reactive process to cigarette smoke in which nonmalignant clonal evolution of LCH cells may occur in the setting of abnormal Langerhans’ cell hyperplasia in the airways.21,22
HISTOPATHOLOGY
The pathologic cell type of pulmonary Langerhans’ cell histiocytosis is the Langerhans’ cell, a differentiated cell of the monocyte-macrophage line (Fig. 61-2). Langerhans’ cells are distinguished by a pale-staining cytoplasm and large convoluted nucleus. Electron microscopy may demonstrate the classic pentalaminar cytoplasmic inclusion known as a Birbeck granule (Fig 61-3). Langerhans’ cells are also characterized by the presence of the CD1a antigen on the cell surface, a feature not found in other cells of histiocytic origin. Langerhans’ cells also react with anti-S-100 antibody, but this reactivity can also be observed in other cell types. More recently, langerin, a type II mannose lectin that is constitutively associated with Birbeck granules, has been identified as a specific marker of Langerhans’ cells.23 Although this cell can be found in association with cigarette smoking in otherwise healthy persons and with other pulmonary pathologies (e.g., idiopathic pulmonary fibrosis) or in normal lung, its presence is characteristic of pulmonary Langerhans’ cell histiocytosis. In pulmonary Langerhans’ cell histiocytosis, the Langerhans’ cells are characteristically found in clusters and significantly outnumber those seen in other lung diseases. Absolute quantitative guidelines for diagnosis of pulmonary Langerhans’ cell histiocytosis have not been established.
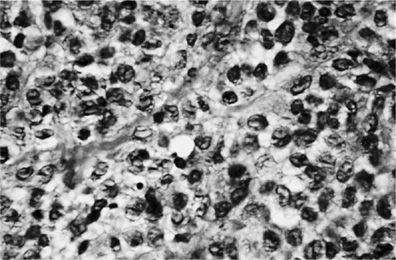
Figure 61-2 Lung tissue in pulmonary Langerhans’ cell histiocytosis. The Langerhans’ cells are typical. A characteristic longitudinal groove is seen along the center of some cells (×96.)
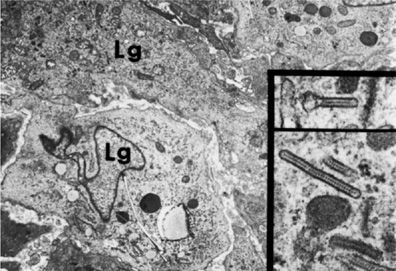
Figure 61-3 Electron micrograph of Langerhans’ cell (Lg) of the lung. Typical X bodies (Birbeck granules) are seen in the two insets.
Early inflammatory lesions center around the smaller bronchioles and usually contain a mixture of Langerhans’ cells, eosinophils, lymphocytes, and neutrophils. The cells appear to invade the bronchiole, destroying the bronchiolar wall in an eccentric fashion. The lesions often affect pulmonary arterioles and venules, so that the disorder can be described as having a bronchovascular distribution.
Pseudodesquamative interstitial pneumonia (characterized by the accumulation of alveolar macrophages in the alveolar parenchyma between pulmonary Langerhans’ cell lesions) and respiratory (smoker’s) bronchiolitis (with pigmented macrophages filling the lumen of bronchioles and the surrounding alveolar spaces) have also often been found on lung biopsy.3,8 In addition, intraluminal fibrosis was often present (86% of specimens). The fibrosis was characterized by mural incorporation, alveolar obliteration, and intraluminal buds. It was mild in extent in 59% of specimens, moderate in 20%, and marked in 9%. These findings support the hypothesis that intraluminal fibrosis serves as a mechanism for alveolar collapse, with progression to interstitial fibrosis and lung remodeling.8
Interstitial fibrosis and small cyst formation with a middle- to upper-zone predominance occur in advancing disease. This middle- to upper-zone predominance differs from that of idiopathic pulmonary fibrosis, which generally has a lower-zone predominance. More advanced lesions extend widely into the lung parenchyma that surrounds the bronchovascular structures and produce the so-called stellate lesions that are characteristic of this disorder.3 Kambouchner et al.24 used three-dimensional reconstructions of serial histologic sections to demonstrate that pulmonary Langerhans’ cell histiocytosis lesions are elongated, sheath-like structures of variable diameter that extend proximally and distally along bronchioles and do not necessarily have a spherical morphology (Fig. 61-4).
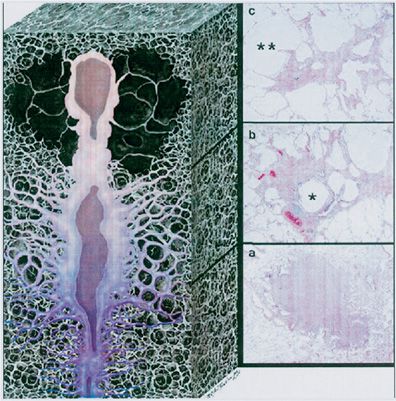
Figure 61-4 Three-dimensional appearance of a pulmonary Langerhans’ cell histiocytosis (PLCH) lesion. Artist’s rendering, based on the reconstructions by Kambouchner et al.,24 illustrates the elongated morphology and variable cellular and fibrotic composition of PLCH with correlative histologic sections. As a PLCH lesion evolves, the nodule of densely packed cells (bottom, A) is centripetally replaced by fibrous tissue and ultimately becomes a stellate scar (top, C). This continuum of change may be evident within a single lesion. PLCH lesions are bronchiolocentric and propagate both proximally and distally along the small airways. The involved bronchiolar lumen may become either dilated or obliterated. The histologic sections correspond to the early, middle, and late phases of PLCH. In the early phase (a), there is a densely cellular nodule with delicate stellate extensions along the adjacent alveolar walls (original magnification, ×12; H&E stain). As the disease progresses (b), cellularity diminishes as fibroblasts replace the lesion (original magnification, ×19.2; H&E stain). Note that the stellate extensions have become more prominent, the central bronchiole (*) is dilated, and adjacent alveolar spaces have coalesced because of focal destruction of alveolar walls (paracicatricial airspace enlargement). In the final phase (c
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


