Essentials of Diagnosis
Pulmonary Hypertension: Introduction
Pulmonary hypertension (PH) is a description of the finding of a mean pulmonary artery pressure (mPAP) greater than 25 mm Hg and may occur in many settings. PH most commonly results from a variety of cardiac and pulmonary diseases that increase pulmonary artery pressure (PAP) either passively or through vasoconstriction. The term pulmonary arterial hypertension (PAH) describes a specific group of diseases characterized by a mPAP greater than 25 mm Hg and normal left heart filling pressures (pulmonary artery wedge pressure [PAWP] < 15 mm Hg) resulting from vasoconstriction and arteriopathy of the precapillary pulmonary arterioles. PAH may be idiopathic or associated with one or more underlying diseases such as connective tissue disease, human immunodeficiency virus (HIV) infection, or portal hypertension. In patients clinically suspected of having PH, echocardiography is often the first test performed to estimate PAP and evaluate right ventricular function. Further comprehensive testing is required to make a diagnosis and establish the etiology of PH.
PH commonly occurs in patients with left heart disease and hypoxemic lung disease. The development of PH in patients with heart or lung disease is an ominous sign and is generally associated with decreased survival. Treatment in this case is directed at the underlying condition (ie, bronchodilators, supplemental oxygen, valve repair, or heart failure treatment). Use of PAH-specific therapy in this setting is generally not beneficial and may worsen symptoms and mortality. In patients with PAH, increased pulmonary vascular resistance from vasoconstriction and arteriopathy results in increased PAP, leading rapidly to right heart failure and death. In this setting, PAH-specific therapy improves symptoms, exercise tolerance, and survival. Selection of therapies for patients with PAH is complex and requires understanding of any associated diseases, the severity of the patient’s symptoms, as well as toxicities and drug interactions of PAH therapy. Early referral to a specialty center for patients with PAH is often crucial for confirmation of diagnosis, early initiation of appropriate therapy, and monitoring of response to treatment. Significant advances have been made over the past decade in improving clinicians’ understanding of the clinical profile, pathobiology, and treatment of PAH.
General Considerations
PH is defined as a sustained elevation in the mPAP of 25 mm Hg or greater at rest. This is in contrast to a normal mPAP of 12–16 mm Hg. In healthy humans, the small arteriovenous pressure gradient generated by blood flow across the pulmonary circulation results from the large total vascular surface area and high pulmonary vascular compliance. The pulmonary vascular resistance (PVR) is quantified using Ohm’s law [PVR = (mPAP – PAWP)/cardiac output (CO)] and describes the relationship between the pressure gradient and blood flow.
Classification & Pathogenesis
The World Health Organization has proposed a system to categorize PH into five groups on the basis of underlying etiology (Table 30–1). PH can also be categorized on a hemodynamic basis at right heart catheterization (Figure 30–1). PH is characterized based on the mPAP: mild (25–40 mm Hg), moderate (41–55 mm Hg), or severe (> 55 mm Hg).
Group 1: Pulmonary arterial hypertension • Idiopathic • Familial • Associated with: Connective tissue disease Congenital systemic-to-pulmonary shunts (including repaired shunts) Portal hypertension HIV infection Drugs and toxins Schistosomiasis Other (thyroid disorders, glycogen storage disease, Gaucher disease, hereditary hemorrhagic telangiectasia, hemoglobinopathies, myeloproliferative disorders, splenectomy) • Associated with significant venous or capillary involvement Pulmonary veno-occlusive disease Pulmonary capillary hemangiomatosis • Persistent pulmonary hypertension of the newborn Group 2: Pulmonary hypertension with left-sided heart disease • Left-sided atrial or ventricular heart disease • Left-sided valvular heart disease Group 3: Pulmonary hypertension associated with lung disease and/or hypoxemia • Chronic obstructive pulmonary disease • Interstitial lung disease • Sleep-disordered breathing • Alveolar hypoventilation disorders • Long-term exposure to high altitude Group 4: Pulmonary hypertension due to chronic thrombotic and/or embolic disease • Chronic thromboembolic pulmonary hypertension • Nonthrombotic pulmonary embolism (tumor, parasites, foreign material) Group 5: Miscellaneous • Sarcoidosis, histiocytosis X, lymphangiomatosis, compression of pulmonary vessels (adenopathy, tumor, fibrosing mediastinitis) |
Figure 30–1.

Hemodynamic classification of pulmonary hypertension (PH). A: Pulmonary hypertension may result from pathology in precapillary or postcapillary pulmonary circulation. B: Common causes of precapillary PH. C: Common causes of postcapillary and mixed PH. Ao, aorta; CMP, cardiomyopathy; LA, left atrium; LV, left ventricle; LVEDP, left ventricular end-diastolic pressure; PA, pulmonary artery; PAWP, pulmonary arterial wedge pressure; PC, pulmonary capillaries; PV, pulmonary veins; PVR, pulmonary vascular resistance; RA, right atrium; RV, right ventricle; VC, vena cava.
Pathologic abnormalities in PAH are localized to the precapillary pulmonary arterioles, resulting in a characteristic hemodynamic profile that can be identified during right heart catheterization (RHC) (see Figure 30–1). PAH is defined by a sustained elevation of the mPAP ≥ 25 mm Hg and a PAWP or left ventricular end-diastolic pressure ≤ 15 mm Hg (normal PAWP, 8–12 mm Hg).
PAH is caused by a disparate group of diseases that have in common vasoconstriction and arteriopathy with remodeling of the precapillary pulmonary arterioles (see Table 30–1). PAH results in progressive cardiopulmonary deterioration, right heart failure, and death. While the prognosis for patients with PAH varies with the underlying cause, overall survival is poor. Figure 30–2 shows survival characteristics in patients with different subtypes of PAH.

The prevalence of PAH in the United States is estimated to be between 50,000 and 100,000 cases. Of these, only 15,000 to 25,000 are appropriately diagnosed and treated. Multicenter registry data suggest that most patients with PAH are women (with a 2:1 female-to-male ratio), with a mean age of 50 years. Seventy-five percent of patients with PAH have symptoms at rest or with minimal exertion. The mean duration of symptoms prior to diagnosis is 27 months.
Three processes contribute to pulmonary artery (PA) luminal narrowing in PAH: vasoconstriction, arterial remodeling, and thrombosis in situ (Figure 30–3). In susceptible individuals, pulmonary vascular injury leads to disruption of the homeostatic balance present in the healthy pulmonary arteries. Upregulation of vasoconstrictive and proproliferative mediators (ie, endothelin-1, serotonin, and thromboxane) and downregulation of vasodilatory and antiproliferative mediators (ie, nitric oxide, prostacyclin, and smooth muscle cell potassium channels) lead to increased vasoconstriction and disordered cell proliferation. In situ thrombus formation occurs as a result of increased platelet activation, increased blood stasis, upregulation of plasminogen activator inhibitor-1, and reduced fibrinolytic activity. The cellular processes that have been implicated in the pathogenesis of PAH are summarized in Figure 30–4.
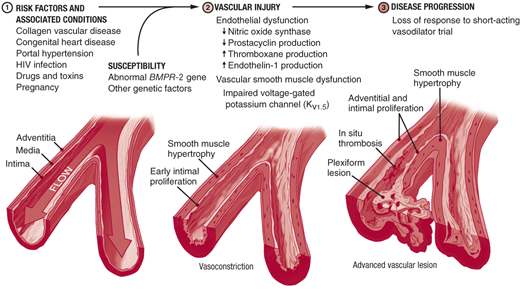
Figure 30–4.
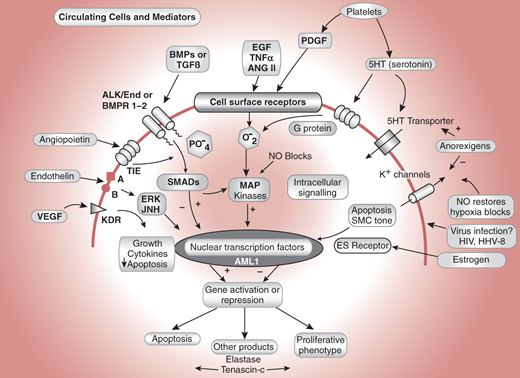
Some cellular processes implicated in the pathogenesis of pulmonary arterial hypertension. VEGF indicates vascular endothelial growth factor; its receptor is KDR. Endothelin is vasoactive and acts through calcium channels and ERK/Jun kinases. TIE is the angiopoietin receptor, a system found to be upregulated in pulmonary vascular disease. Alk1 and BMPR1-2 are TGF receptors. Alk1 mutations cause hereditary hemorrhagic telangiectasia and some cases of pulmonary hypertension. Epidermal growth factor, tumor necrosis factor, angiotensin II, and platelet-derived growth factor are all proliferative stimuli that act through tyrosine kinase receptors. SMADs are regulatory proteins that activate nuclear transcription factors and interact with MAP kinases. AML1 is a nuclear transcription factor. (Reprinted, with permission, from Newman JH. Circulation. 2004;109:2947.)
PAH is termed idiopathic if no disease associated with PAH is present and heritable if there is an underlying genetic predisposition or familial occurrence of PAH. Idiopathic and heritable PAH are rare disorders with a prevalence of 15 cases per million per year and an incidence of 1–2 cases per million per year. Idiopathic and heritable PAH afflict predominantly young women, with a mean age at diagnosis of 35 years. These are progressive disorders with a high mortality (median life expectancy 2.8 years from diagnosis).
Mutations in the bone morphogenetic protein receptor (BMPR)-2 gene have been identified in approximately 50% of patients with heritable PAH. Those who inherit the mutation have a 10% risk of developing PAH. The genetic pattern of inheritance is autosomal dominant with incomplete penetrance and anticipation (subsequent generations manifest the disease at an earlier age). A mutation in the activin receptor-like kinase type-1 (ALK-1 or endoglin) often with coexistent hereditary hemorrhagic telangiectasia may have also been identified in patients with PAH. Both BMPR-2 and ALK-1 are members of the transforming growth factor β signaling pathway.
Diseases associated with PAH include collagen vascular diseases (especially systemic sclerosis), congenital systemic-to-pulmonary shunts, portal hypertension, HIV infection, drug (notably anorexigens) and toxin exposures, and other disorders as delineated in Table 30–1.
For those patients in whom PAH develops secondary to collagen-vascular disease, the prognosis is extremely poor. In patients with PAH associated with systemic sclerosis, for instance, the 1-year survival is approximately 50%.
Congenital systemic-to-pulmonary shunts result from ventricular or atrial septal defects, anomalous pulmonary venous drainage, patent ductus arteriosus, or an aorto- pulmonary window. Systemic-to-pulmonary shunts expose the pulmonary vasculature to a persistent high-flow state. This can lead to PA endothelial dysfunction, mediator activation, and reactive vasoconstriction. Vascular remodeling ensues and can progress to the point that shunt reversal occurs (Eisenmenger syndrome). Shunt reversal worsens hypoxemia and further exacerbates PAH. PAH may also develop many years after repair of a congenital shunt as a long-term consequence of vascular injury sustained prior to shunt repair.
PAH develops in approximately 5–10% of patients with portal hypertension. Porto-pulmonary hypertension can interfere with eligibility for liver transplantation because it is associated with high perioperative mortality.
It is estimated that PAH develops in 1 of every 200 patients with HIV infection. Those patients with HIV in whom PAH develops have a particularly poor prognosis (see Figure 30–2). Methamphetamine use is also associated with the development of PAH. The diet drug fenfluramine, which has been removed from the U.S. market, has also been associated with increased likelihood of PAH.
The disorders discussed thus far, while distinct, result in pulmonary endothelial and vascular smooth muscle cell dysfunction, maladaptive arterial remodeling, and increased vascular resistance. While prognosis of PAH varies based on the underlying disease, presence of PAH significantly increases the morbidity and mortality of all associated conditions.
Elevation of left-sided cardiac filling pressures can lead to PH through passive congestion and retrograde transmission of elevated pressure to the PA tree. This is termed “postcapillary pulmonary hypertension” (see Figure 30–1A, C). Postcapillary PH is characterized hemodynamically by an mPAP ≥ 25 mm Hg, PAWP > 15 mm Hg, and a normal PVR. Elevation of left-sided cardiac filling pressures may result from aortic or mitral valve dysfunction, myocardial disease, pericardial disease, atrial myxoma with obstruction, or pulmonary vein compression. With chronic elevation of pulmonary venous pressures, upregulation of inflammatory cytokines, as well as vasoconstrictive and proproliferative mediators, results in reactive vasoconstriction. Elevation of PA pressure can therefore exist out of proportion to left-sided filling pressures. This entity is referred to as mixed PH or “reactive PH” and is characterized hemodynamically by an mPAP ≥ 25 mm Hg, PAWP > 15 mm Hg, and a PVR > 3 Wood units (240 dynes × s × cm−5).
Since the PVR is normal in pure postcapillary PH, optimal management of left-sided heart disease with appropriate therapies that reduce the left-sided cardiac filling pressure (ie, surgery for valvular dysfunction, diuretics, angiotensin-converting enzyme inhibitors, hydralazine) will result in normalization of the PAP. This may not be the case if mixed PH has developed, as is seen in patients with severe end-stage heart failure undergoing transplant evaluation. Targeted vasodilator therapy can often improve reactive PA vasoconstriction in this setting. However, “fixed” mixed PH that is refractory to vasodilator therapy from vascular arterial remodeling can pose a significant problem in patients undergoing heart transplantation. Acute right heart failure may occur when an unconditioned donor right ventricle is exposed to irreversibly high PVR after transplantation.
PH is also associated with diseases that affect the lung parenchyma such as chronic obstructive pulmonary disease (COPD) and interstitial lung disease (ILD). PH secondary to lung disease is hemodynamically precapillary in origin (see Figure 30–1A, B). Several factors contribute to the development of PH in this setting. Disruption of capillary networks results in a decrease in the overall surface area of the pulmonary vascular bed and thereby increases PVR. Hypoxemia directly triggers pulmonary vasoconstriction and indirectly contributes to pulmonary vascular remodeling by resulting in upregulation of proproliferative mediators. Hyperviscosity secondary to hypoxia-induced erythrocytosis can worsen PH. Sleep-disordered breathing, alveolar hypoventilation disorders, and prolonged exposure to high altitude are other causes of PH associated with hypoxemia.
Treatment of these disorders is based on correction of the underlying disorder, provision of supplemental oxygen, and management of cor pulmonale. Although some benefits from PAH-specific therapy have been observed in ILD patients, these treatments also worsen hypoxemia because they inhibit physiologic pulmonary vasoconstriction and worsen ventilation-perfusion mismatch. Use of PAH-specific therapy in this setting is generally discouraged.
A massive, sudden pulmonary embolus (PE) may significantly increase PAP and cause hemodynamic collapse secondary to acute right ventricular failure. This scenario leads to death in approximately 20–40% of patients with acute PE. The presence of severe PH in this setting portends a poor prognosis and predicts an increased likelihood of chronic thromboembolic PH (CTEPH). In fact, most patients with pulmonary embolism experience complete resolution of thromboemboli within 30 days.
CTEPH occurs in up to 4% of patients following acute PE or 10% following recurrent PE. However 50% of patients diagnosed with CTEPH have no history of a thromboembolic event. Why some patients develop CTEPH is not well understood, but surgical specimens show evidence of persistence and organization of thromboembolic material as well as vascular remodeling characterized by intimal thickening with deposition of collagen and hemosiderin, atherosclerosis, and calcification. Vasculopathy and pathologic remodeling of unobstructed downstream pulmonary arterioles similar to that which occurs in idiopathic PAH also develops possibly secondary to abnormal blood flow.
Mortality in patients with CTEPH is high but, in selected patients, can be improved markedly with surgical pulmonary thromboendarterectomy (PTE). Excluding CTEPH as a cause of PH requires evaluation with a ventilation-perfusion scan followed by confirmatory conventional pulmonary angiography. Computed tomography (CT) pulmonary angiography is not adequately sensitive to definitively exclude the diagnosis. Early referral to a center that can perform PTE is needed.
This category of PH encompasses a diverse group of disorders that are hemodynamically precapillary in origin and includes sarcoidosis, histiocytosis X, lymphangiomatosis, and pulmonary vein compression.
Elevations in PAP eventually result in right ventricular systolic failure from chronic pressure overload (Figure 30–5). In response, the right ventricle undergoes hypertrophic remodeling to maintain CO and compensate for increased afterload. Early on, the right ventricle can have supernormal function and normal-to-reduced chamber dimensions. Chronic right ventricular pressure overload results in persistent upregulation of proproliferative neurohormones, endothelin-1, and cytokines. These mediators contribute to the development of maladaptive hypertrophy with fibrosis and diastolic dysfunction.
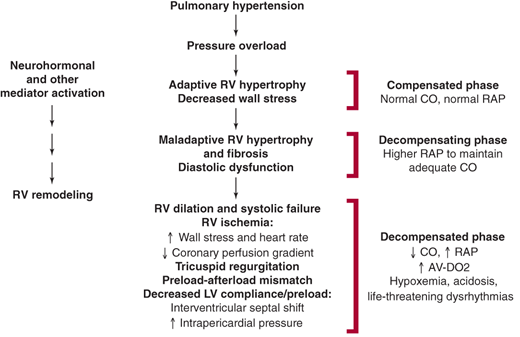
If exposure to elevated PA pressure continues, the right ventricle dilates. This is an ominous sign because it signifies the presence of increased right ventricular wall stress. Increased wall stress, when coupled with increased heart rate, results in increased myocardial oxygen demand. This develops in concert with a reduction in the epicardial-to-endocardial coronary perfusion gradient secondary to increased right ventricular end-diastolic pressure. In sum, these changes result in a myocardial oxygen supply-demand mismatch and right ventricular ischemia. Right ventricular ischemia worsens systolic function, increases end-diastolic pressure, and promotes further ventricular enlargement. Tricuspid regurgitation secondary to annular dilatation often occurs and serves to further reduce effective right ventricular forward output.
Right ventricular dilation, in the setting of an intact pericardium, results in a shift of the interventricular septum toward the left ventricle. This, especially when coupled with increased intrapericardial pressure, impedes left ventricular filling (preload). In turn, when left ventricular filling is impaired, systemic CO is reduced.
Multiple mechanisms contribute to the development of hypoxemia in PH, even when intrinsic pulmonary disease (COPD, CTEPH) is absent. Pulmonary vascular remodeling and in situ thromboses disrupt normal capillary-alveolar gas exchange and raise the alveolar-to-arterial oxygen gradient. Increased pulmonary pressures can increase right atrial pressure and cause shunting through a patent foramen ovale. Because a patent foramen ovale is present in up to 30% of adults, right-to-left shunting should be considered in patients with PH and hypoxemia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


