HRCT technical requirements
Scans without contrast
Full inspiration without respiratory motion
Contiguous or noncontiguous axial scans with thin sections, reconstructed at ≤2 cm intervals
Reconstructed slice collimation ≤2 mm
High resolution reconstruction algorithm
Field of view to include lungs only
Expiratory scans, to exclude lobular air trapping suggestive of hypersensitivity pneumonitis
Prone scans if dependent density obscures detail on supine images
Optional coronal and sagittal reconstructions if volumetric images are obtained
According to these guidelines, a definite UIP pattern on chest HRCT can be identified when all the following features are present: (1) subpleural, basal predominance; (2) reticular abnormalities; (3) honeycombing with or without traction bronchiectasis and (4) absence of features that are inconsistent with a UIP pattern (such as any of the following: upper or mid-lung predominance, peribronchovascular predominance, extensive ground glass abnormality, profuse micronodules, discrete cysts away from areas of honeycombing, diffuse mosaic attenuation/air trapping, segmental or lobar consolidation) [4]. The presence of features one, two and four in the absence of honeycomb changes defines a possible UIP pattern, which should require confirmation with SLB in order to achieve a final diagnosis.
Therefore, a HRCT UIP pattern should be considered in patients who present with low lung volumes, subpleural reticular opacities, honeycombing with or without traction bronchiectasis, the extent of which increases from the apex to the bases of the lungs. In a patient with a definite UIP pattern on chest HRCT, the disease is most extensive on lower sections. Typically, imaging findings of a UIP pattern are heterogeneous, with areas of more or less dense fibrosis alternating with areas of normal parenchyma. The dominant finding is represented by honeycombing, a critical element for the definition of definite UIP on HRCT. According to the definition of the Fleischner Society, honeycombing consists of clustered cystic air spaces typically of comparable diameters in the order of 3–10 mm, but occasionally as large as 2.5 cm. It usually has a subpleural distribution with well-defined shared walls [6]. The finding of honeycombing generally implies the presence of established (and therefore irreversible) lung fibrosis.
Although the presence of honeycombing makes an important difference in identifying a definite UIP pattern according to current guidelines, more recent data might question the ability of even expert radiologists of agreeing in the identification of such a finding on HRCT [7]. The presence of traction bronchiectasis (which are also a critical component in the radiological definition of the UIP pattern) or superimposed emphysema represents major causes of disagreement, with resulting low diagnostic agreement. Multiplanar image reconstruction might be helpful in distinguishing definite honeycombing from its mimics, particularly from traction bronchiectasis or bronchiolectasis in the context of surrounding extensive pulmonary fibrosis. As per authors’ admission, the differentiation between true honeycombing and traction bronchiectasis might be subjective, and the presence of a combination of both is also a possibility, making the distinction virtually impossible [7]. However, whether different imaging criteria for the definition of honeycombing have any impact on patients’ prognosis is presently unknown and requires further investigation, particularly in the context of more recent data suggesting that traction bronchiectasis are critical elements in predicting clinical course in ILDs.
On the other hand, we know that the positive predictive value of a HRCT diagnosis of UIP is as good as up to 90–100 % [8–13] when compared with SLB pathology, therefore a UIP pattern on HRCT is highly accurate for the presence of UIP pattern on surgical lung biopsy. If honeycombing is absent, but the imaging features otherwise meet criteria for UIP, the imaging features are regarded as representing possible UIP, and, if clinically indicated, SLB is recommended to increase diagnostic accuracy. A recent assessment of HRCT images from patients screened for the inclusion in an IPF randomized control trial has demonstrated that, in the proper clinical setting and in the context of a high radiological expertise in ILDs, even a possible UIP pattern on HRCT can be predictive of a definite UIP pattern on histology. These data would suggest that, upon careful assessment of each individual case by expert clinicians and radiologists, surgical lung biopsy might be avoided in some cases, in which HRCT demonstrates a possible UIP pattern [14]. Further confirmation of these data in other cohorts would be crucially important. Moreover, it is relevant to remember that even in patients whose chest HRCT doesn’t fit with a UIP pattern, a SLB may still show UIP pattern on histopathology.
Ground-glass opacities can be present in many patients with a UIP pattern but in the context of IPF usually they are less prevalent than honeycombing and reticular abnormalities [15–17]. If ground-glass opacities predominate, an extensive search for a diagnosis other than IPF should be undertaken. Similarly, the presence of micronodules, air trapping, non-honeycomb cysts, consolidation, or a peribronchovascular-predominant distribution should lead to consideration of an alternative diagnosis [18, 19]. Chest radiograph is less useful than HRCT in evaluating patients with suspected IPF [8], and in fact it can be completely normal in patients with early disease. In advanced disease, the chest radiograph shows non specific decreased lung volumes and subpleural reticular opacities that increase from the apex to the bases of the lungs [20].
The Pathologist’s View: UIP Pattern on Lung Biopsy
The morphologic diagnosis of UIP pattern is no longer the gold standard in IPF, since (as outlined above) in recent years HRCT has provided reproducible information in confirming such a diagnosis in the majority of cases. In other words, accuracy of HRCT in a typical case of IPF is almost perfect and histology is not necessary. However, histology still has a role when HRCT is not typical for a definite UIP pattern, since the differential diagnosis between IPF and NSIP has important prognostic and therapeutic consequences.
On histology, the UIP pattern is a non uniform fibrotic process with subpleural and paraseptal scarred fibrosis irregularly and abruptly juxtaposed to normal lung (spatial heterogeneity with patchwork pattern) (Fig. 21.1a) and this finding is particularly evident at low magnification. Lung architecture is distorted with honeycomb changes (Fig. 21.1b) characterized by enlarged airspaces lined by metaplastic bronchiolar-type epithelium and filled with mucus, neutrophils, macrophages and/or giant cells with intracytoplasmic cholesterol cleft. Seminal papers have highlighted the most important features characterizing a UIP pattern on histology [21–23]. All expert pulmonary pathologists agreed in recognizing three key features: (1) patchy involvement of lung parenchyma consisting of alternating areas of scarred and normal tissues; (2) architectural distortion with/without honeycombing appearance; and (3) the finding of active fibroblastic foci abruptly adjacent to old, dense fibrosis. In advanced/end stage UIP pattern, the lung architecture is completed obscured by diffuse honeycombing and smooth-muscle scars without interposed normal lung. The hallmark of the active fibrotic process is represented by fibroblastic foci (Fig. 21.1c), consisting of dome-shaped proliferation of myofibroblasts into a myxoid stroma, often covered by hyperplastic pneumocytes or bronchiolar cells. The “young” active fibrosis is easily appreciated as small spots of “gray-to-blue” hematoxylinophilic colour into an “old” remodelling fibrosis with a typical “pink” eosinophilic colour. This peculiar alternation of “old” (fibrotic scars and honeycombing) and “young” (fibroblastic foci) fibrosis leads to the concept of temporal heterogeneity (Fig. 21.1d).
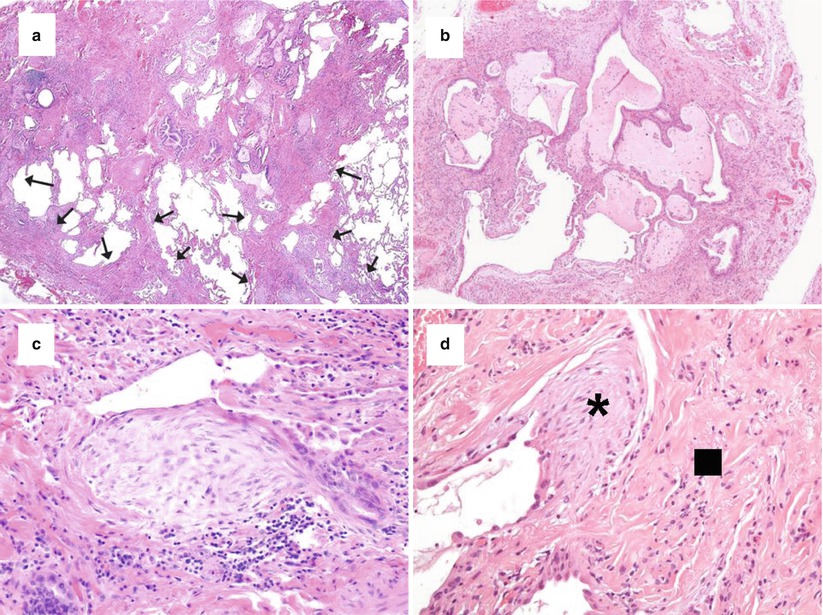
Fig. 21.1
Histologic features of UIP. The patchwork pattern characterized by scarred fibrosis (arrows) abruptly alternated by normal lung with main involvement of peripheral subpleural areas and paraseptal regions (a, haematoxylin-eosin × 40). Honeycomb changes with enlarged airspaces filled by mucus and inflammatory cells and surrounded by scarred fibrosis and lined by bronchiolar-type epithelium (b, haematoxylin-eosin × 100). Fibroblast focus representing the active phase of fibrosis with longitudinally-oriented myofibroblasts in a myxoid matrix covered by hyperplastic pneumocytes or bronchiolar epithelium (c, haematoxylin-eosin × 200). Active (asterisk) and old fibrosis (square) abruptly alternated (d, haematoxylin-eosin × 200)
Two concepts that have been stressed by the evidence-based IPF guidelines [4] concern the level of confidence for a diagnosis of UIP and the introduction of exclusion criteria for UIP. Four main histopathologic criteria have been identified: (1) architecture distortion by scarred fibrosis with or without honeycombing; (2) patchwork pattern; (3) fibroblastic foci; (4) lack of features excluding UIP pattern. Based on these features, four categories/levels of confidence have been proposed: (a) definite UIP, which requires the presence of all four criteria; (b) probable UIP, requiring the presence of features one and four, or the presence of honeycomb changes only due to an end-stage fibrotic disease; (c) possible UIP, requiring the presence of features two and four; (d) not UIP, due to the presence of several histologic features (exclusion criteria) which stands against a UIP pattern, such as hyaline membranes (Fig. 21.2a), granulomas (Fig. 21.2b), organizing pneumonia (Fig. 21.2c), a marked inflammatory infiltrate away from honeycombing (Fig. 21.2d), airway-centred changes, other features suggesting an alternative diagnosis (e.g. asbestos bodies) (Table 21.2).
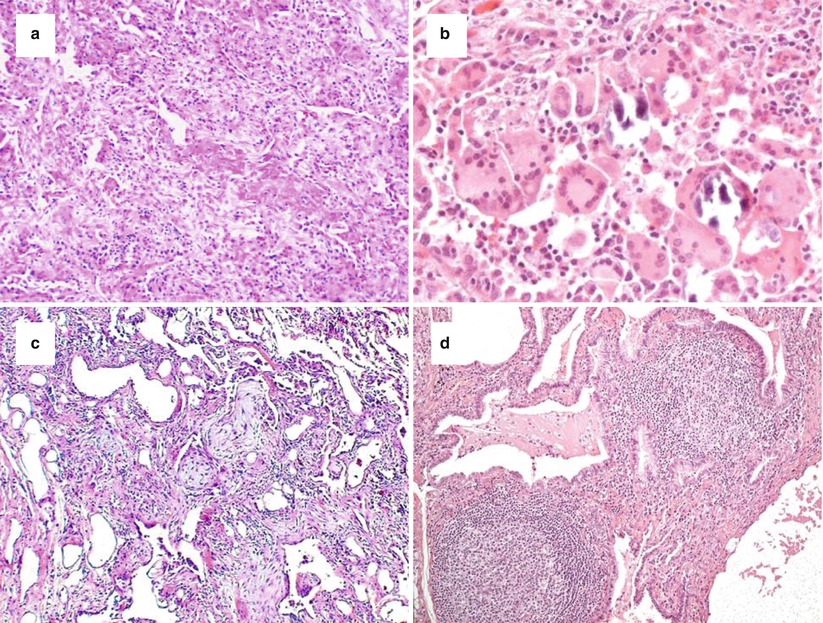
Fig. 21.2
Morphologic features excluding a UIP pattern, such as hyaline membranes (a, haematoxylin-eosin × 100), granulomatous inflammation ± giant cells (b, haematoxylin-eosin × 200), organizing pneumonia (c, haematoxylin-eosin × 100), and marked inflammatory infiltrate (d, haematoxylin-eosin × 200)
Definite UIP | Probable UIP | Possible UIP | Not UIP |
|---|---|---|---|
1 + 2 + 3 + 4 | 1 + 4 or Honeycombing only (end-stage lung) | 2 + 4 | 5 |
Histologic examination for the evaluation of the presence of a UIP pattern is usually performed on SLB. As such, the pathological criteria for the definition of a UIP pattern, proposed in the 2011 evidence-based IPF guidelines [4], apply only to SLB samples. The ability to select different sites (biopsies should be taken from at least two different lobes) with deep and large biopsies based on a previous accurate HRCT analysis is mandatory to prevent sampling errors and misleading diagnoses [24, 25]. IPF is heterogeneous, often affecting people with smoking history; therefore the finding of areas showing an NSIP pattern or smoking-related changes along with features of UIP should be taken in account. However, prognosis is basically related to the finding of UIP, even when UIP pattern represents the minor fibrotic component [26–28].
While SLB remains the standard technique for identifying a UIP pattern on histology, some preliminary results indicate a possible role of transbronchial lung biopsy (TBLB) [29]. In a recently published retrospective experience [30], the authors evidenced that histopathologic criteria to suggest a UIP pattern (i.e. at least one feature among patchy interstitial fibrosis, fibroblast foci, honeycomb changes) were present in 30 % of all UIP cases. Sensitivity increased with a higher number of biopsies and the size of specimens, and the agreement between two expert pathologists was good (kappa = 0.61). However, TBLB may be performed only in selected cases and further prospective studies are needed in order to assess the actual sensitivity and specificity of TBLB for the diagnosis of a UIP pattern.
On the other hand, despite a representative tissue sampling, another possible limitation in recognising a UIP pattern on histology is related to the great inter-observer variability among pathologists (mirroring what was described above for chest radiologists). Several studies have highlighted that inter-observer agreement is better, but far from being perfect, among expert pathologists as compared to non-expert pathologists, and very low when comparing experts and non-experts [31–33].
From Patterns to Clinical Diagnosis
The UIP Pattern in IPF
The 2011 evidence-based IPF guidelines [4], and the previously published documents such as the Consensus Statement for Classification of Idiopathic Interstitial Pneumonias (IIP) [3], provide a common framework to interpret radiology and pathology features in terms of patterns and to translate patterns into clinical diagnoses of an idiopathic ILD. In the proper clinical setting, the demonstration of a definite UIP pattern at chest HRCT and/or at SLB is considered diagnostic for IPF, the ILD with the worse prognosis. Therefore, the first request that clinicians address to radiologists and pathologists is to differentiate a UIP pattern from non-UIP pattern, since, in the absence of secondary causes of ILD, the recognition of a UIP pattern leads to the clinical diagnosis of IPF. By combining radiologic patterns with histology patterns, the guidelines on IPF diagnosis and management have proposed a scheme to assign a diagnosis of IPF according to a scale consisting of different degrees of confidence [4]. While it is allowed to attribute a diagnosis of IPF based on a definite UIP pattern on HRCT in the absence of histology confirmation, given the high positive predictive value of HRCT, it has been recognized that, upon multidisciplinary discussion, IPF may be diagnosed also on biopsy-proven UIP in an asymptomatic patient lacking imaging features of UIP, in the effort to recognize and treat patients with early disease.
It is important to state for the pathologists (and clinicians too) that the diagnostic classification according to the levels of confidence in the final diagnosis and the terminology proposed in the 2011 IPF guidelines [4] are important and useful in allocation of cases in clinical trials, while labelling a UIP pattern and therefore a diagnosis of IPF with a level of confidence may not be feasible or even appropriate in the routine practice. In fact, these levels of confidence, mostly derived from retrospective studies, have not been yet validated and basically have a conceptual value in indicating how the pathologist is certain about a UIP pattern rather than being of practical use.
The UIP Pattern in Other ILDs
A UIP pattern on HRCT and/or histology does not always lead to a clinical diagnosis of IPF. In fact, it should be reminded that similar radiologic or pathologic patterns may be seen in idiopathic and non-idiopathic interstitial pneumonias, therefore the simple recognition of any given pattern does not exempt from a careful evaluation for possible secondary causes of ILD. For example, a UIP pattern on HRCT is not exclusively seen in IPF, as it can be the distinctive feature also in the case of lung involvement in connective-tissue disease (CTD), particularly in rheumatoid arthritis-associated interstitial lung disease [34], in chronic hypersensitivity pneumonitis (HP) [35], or in mineral dust-related interstitial lung disease, such as asbestosis [36]. Despite a great extent of overlap in UIP pattern from different clinical entities, some peculiar features might be helpful in suggesting alternative diagnosis, particularly when chronic HP and CTD-ILD can be suspected [37]. For example, the presence of lobular areas of decreased attenuation and centrilobular nodules, along with the absence of distinct lower zone predominance of abnormalities, appear to be helpful in differentiating IPF from chronic HP on HRCT in approximately 50 % of patients [38], although the causative antigen might remain unknown in a sizeable proportion of patients with histology-proven chronic HP [39]. The presence of ground-glass opacities, non-honeycomb cysts or consolidation, even on an otherwise UIP-like background, should suggest alternative diagnoses other than IPF and possibly require histology confirmation. The coexistence of pleural or pericardial effusion, a dilated oesophagus or airway-associated abnormalities might be evocative of CTD-ILD [37]. Serologic testing for CTD (including rheumatoid factor, anti-cyclic citrullinated peptide and anti-nuclear antibody titre and pattern) is recommended in the majority of patients in diagnostic work-up of suspected IPF and certainly a careful assessment should be performed in younger female patients (less than 50 years of age), possibly presenting with some features of CTD such as Raynaud phenomenon, sicca syndrome or arthritis [4, 40]. Furthermore, despite a similar UIP pattern, patients presenting with a CTD-related ILD have a better prognosis as compared to patients with IPF [40, 41].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


