6

Pulmonary Disorders in the
Neonate, Infant, and Child
This chapter discusses lower respiratory tract disorders encountered in the pediatric age group. They encompass a broad spectrum of individually uncommon conditions including congenital anomalies, disorders of lung growth, and developmental and acquired pulmonary vascular abnormalities of importance in this age group. The more recently characterized abnormalities of the surfactant system related to surfactant dysfunction mutations are considered in some detail. Disorders of the transition from intra- to extrauterine life with their short- and long-term respiratory complications are also covered, as are some iatrogenic problems and a few specific infections of clinical importance in infants. The pulmonary manifestations of cystic fibrosis and other metabolic and genetic diseases that affect the lung are briefly outlined. Neoplasms of the respiratory tract are rare in infancy and childhood, and the majority are metastatic; the few rare primary neoplasms of the lung that occur in childhood are considered.
Developmental Disorders of the Lower Respiratory Tract
Developmental disorders of the lung, except for those related to abnormalities of septation of the laryngotracheal groove, are poorly understood in terms of the embryologic mechanisms involved. There is, however, emerging evidence that surfactant protein genes, extra-cellular matrix molecules, and a variety of growth factors and their receptors are important in both structural development and differentiation in the embryonic and fetal lung. This is likely to lead to a greater understanding of disorders of development as such systems can be experimentally perturbed.
Pulmonary Agenesis
Bilateral Pulmonary Agenesis
There are rare reports of bilateral congenital absence of the lung.1–4 In this abnormality the respiratory tract distal to the trachea fails to develop. Most cases have associated abnormalities of tracheal or esophageal development, particularly tracheal agenesis or esophageal atresia, and there also may be asplenia. Evidence suggests that the underlying mechanism for such abnormalities may be a defect in the expression of fibroblast growth factor or its receptors in the developing lung bud.5
Unilateral Pulmonary Agenesis
Unilateral agenesis of the lung is more common than bilateral.6–8 It is, however, a rare disorder. It has a wide spectrum of presentation from asymptomatic cases discovered incidentally to respiratory distress in the newborn.9 It may be compatible with essentially normal life, particularly if there are no other anomalies; however, there are often other associated congenital anomalies.9,10 Absence of the left lung is thought to have a better prognosis than right pulmonary agenesis. The excess morbidity of the latter may be related to tracheal or bronchial compression by the deviated aorta (Fig. 6–1) or other mechanical effects related to the often severe mediastinal shift that can accompany unilateral agenesis. Unilateral pulmonary agenesis may be associated with a variety of other developmental malformations including facial abnormalities; skeletal changes involving the vertebrae, distal limbs, and chest wall; tracheal agenesis and bronchoesophageal fistula; and several cardiovascular abnormalities, particularly septal defects and anomalous pulmonary venous connection.9,11,12 Many of the changes seen with unilateral pulmonary agenesis are also components of the VACTERL (vertebral abnormalities, anal atresia, cardiac abnormalities, tracheoesophageal fistula or esophageal atresia, renal agenesis and dysplasia, and limb defects) association, and it has been suggested that unilateral pulmonary agenesis may fall within that constellation of abnormalities.13 There also appears to be a particular association with ipsilateral radial ray defects or hemifacial microsomia.14 Unilateral agenesis has been described as a component of Goldenhar’s syndrome, characterized by facial asymmetry, external ear malformations, conductive hearing loss, eye malformations, vertebral anomalies, aplasia or hypoplasia of the mandibular ramus, and various renal anomalies. It has been associated with nonimmune hydrops.15 There is an increased incidence of patent ductus arteriosus.7
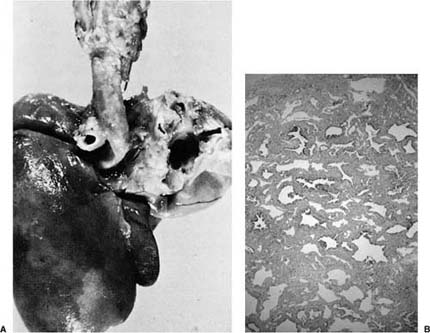
FIGURE 6–1 (A) Agenesis of the right lung, posterior view. Note the prominent aortic indentation in the left main-stem bronchus. The patient had clinical evidence of airflow obstruction. (B) The small amount of residual left lung tissue from a 3-year-old boy with left pulmonary agenesis shows marked parenchymal changes with irregular simplified airspaces and airways distributed in abundant matrix. (Courtesy of Dr. K. Lancaster.)
In unilateral agenesis (Table 6–1), the trachea may extend directly and smoothly into the single main bronchus (group A), or a rudimentary bronchus to the absent lung may be present (group B). Sometimes there may be maldeveloped lung tissue as well (group C) (Fig. 6–1). This latter group is the least common form of unilateral agenesis, and is sometimes considered to be a form of hypoplasia. The pulmonary artery and veins are absent or sometimes hypoplastic on the affected side in unilateral pulmonary agenesis.16 Morphometric analysis in one case of unilateral agenesis revealed an increase in volume of the single lung with an increased number of smaller than normal alveoli; the number of bronchial branches was reported to be reduced.8 These changes have some similarities to compensatory alveolar development and may be related to the increased space available to the single lung during in utero development.
Bilateral agenesis: Absence of bronchi, gas-exchanging tissue, and pulmonary vasculature except for main pulmonary artery, sometimes absent trachea |
Unilateral agenesis |
Group A: Complete absence of the airways, gas-exchanging tissue, and vasculature of the affected lung |
Group B: Rudimentary main-stem bronchus, but no other airways or gas-exchanging tissue, vasculature absent or hypoplastic |
Group C: Rudimentary bronchus with a small amount of associated maldeveloped lung tissue, vasculature hypoplastic |
Lobar agenesis of the right middle lobe and left lower lobe |
Modified from Gould SJ, Haselton PS. Congenital abnormalities. In: Haselton PS, ed. Spencer’s Pathology of the Lung, 5th ed. New York: McGraw-Hill, 1996:86–87.
Unilateral pulmonary agenesis may be completely asymptomatic. Some affected individuals present with only tachypnea in infancy, whereas others present with recurrent pneumonia, particularly those with a group B lesion where the rudimentary bronchus may be a focus of infection for the single lung.11
Lobar Agenesis
Isolated absence of one or more lung lobes is rare. There are reports of isolated absence of the right middle lobe.11 Absence of the left lower lobe has been reported in association with stenosis of the left main bronchus. Lobar agenesis should not be confused with the far more common abnormalities of bronchial branching and lobation.
Abnormalities of Lobation and Bronchial Branching
Most abnormalities of lobation and bronchial branching are of no clinical significance. Abnormalities of lobation are common and should generally be thought of as normal variants. These include both increased lobation with extra fissures, such as those seen with azygos lobe and cardiac lobe, and absence of major fissures. Azygos lobe is found on the medial surface of the right upper lobe and is demarcated by a fissural depression made by the azygos vein as it courses through this region. Cardiac lobe is also seen in the right lung when the anterior basal segment of the right lower lobe is separated from the remainder of the lower lobe by a fissure. More commonly there may be a reduction in the number of fissures, and typically unilobar left lung or bilobed right lung with absence of the transverse fissure. These abnormalities of lobation have no clinical significance, although they may occasionally be associated with abnormalities of other organ systems.
Occasionally fissural abnormalities may be acquired related to abnormal ventilatory patterns during post-neonatal lung growth, and to the bronchiolar obliteration occasionally seen in chronic neonatal lung disease, particularly in immature infants. These fixed abnormalities of ventilation with atelectasis of lobules and overexpansion of adjacent lobules results in accessory fissure formation, which has been a prominent feature in long-term survivors of bronchopulmonary dysplasia (see Chronic Neonatal Lung Disease, later in the chapter) (Fig. 6–2).
Abnormalities of bronchial origin and branching, similar to those of external lobation, are usually incidental findings of little or no clinical significance.17 These abnormalities include both supernumerary bronchi and less common reductions in major bronchi. Both of these conditions are quite rare. Minor abnormalities in bronchial origin and branching pattern are relatively common and may involve any lobe. Major abnormalities are more likely to involve the right upper lobe.
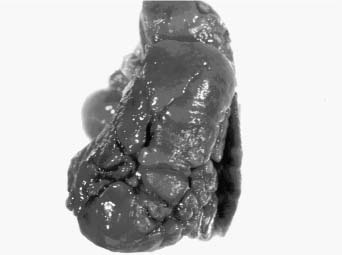
FIGURE 6–2 This lung from a 6-month-old infant with bronchopulmonary dysplasia shows the multiple accessory fissures that may be seen in that disorder.
Tracheal Bronchus
Displaced or ectopic bronchial origin is encountered with some frequency; it is said to be present in 2% of children requiring bronchoscopy for respiratory symptoms.18,19 Although the term tracheal bronchus has been applied to several abnormal bronchial branching patterns, all have in common the origin of a bronchus directly from the trachea. Some types of tracheal bronchus are vestigial tracheal diverticulum, high apical lobe, and supernumerary aerated tracheal bronchus; however, the most usual is a displaced right upper lobe bronchus arising at the junction of the middle and distal thirds of the right lateral trachea.20 It is more common in males and may be associated with other congenital anomalies, as well as right main bronchus stenosis. Abnormalities of bronchial origin usually produce no symptoms. Atelectasis may be seen in intubated patients, others may more stridor, cough, and recurrent right-sided pneumonia.18–20
Pulmonary Isomerism
Abnormalities of external lobation generally do not reflect the pattern of bronchial branching, except in those situations in which an abnormal pattern of bronchial branching leads to similar-appearing right and left lungs. These, either bilateral bilobed “left” lungs or bilateral trilobed “right” lungs, are likely to be associated with significant abnormalities in other systems, particularly the cardiovascular. Bilateral right lungs or right pulmonary isomerism is a cardinal feature of the Ivemark or asplenia syndrome and M-anisosplenia with their associated cardiac defects. The “bilateral” left lungs of left pulmonary isomerism polysplenia and F- and O-anisosplenia syndromes also have associated cardiac defects. These syndromes have now been grouped into the broader category of heterotaxy, where abnormalities of sidedness are known to be genetic in origin. Several genes related to left-right axis abnormalities have been implicated.21
Bridging Bronchus
With the so-called bridging bronchus, the trachea divides into two branches: the right main-stem bronchus gives rise to right upper and middle lobe bronchi, whereas the right lower lobe bronchus arises from the left main-stem bronchus and courses across the mediastinum to the right lower lobe. In the few reported cases the right lung is bilobed, and areas of airway stenosis are seen. This anomalous branching pattern may be seen in association with type 2 left pulmonary artery sling and horseshoe lung.22 There are also several other anomalous bronchi and variations of bronchial branching that have been described. These include the accessory cardiac bronchus arising from the bronchus intermedius, origin of the anterior segmental upper lobe bronchus from the middle lobe bronchus or posteparterial bronchus, and many minor variations.
Main-stem, lobar, or segmental bronchi may abnormally connect with foregut structures, the esophagus or stomach, as part of complex bronchopulmonary foregut malformations. In these situations there may occasionally be associated abnormalities of pulmonary arterial supply or venous connection.
Positional Abnormalities of the Lung
Scimitar Syndrome, Horseshoe Lung, and Crossover Lung Segment
Scimitar syndrome, horseshoe lung, and crossover lung segment are related abnormalities. Scimitar syndrome is a rare developmental cardiopulmonary anomaly in which total or partial anomalous pulmonary venous connection of the right lung to the inferior vena cava is associated with right lung hypoplasia, dextroposition of the heart, and anomalous systemic arterial supply to the right lung. The scimitar-like chest x-ray appearance of the anomalous venous drainage gives the syndrome its name. Within this syndrome there are multiple variant forms of pulmonary venous connection and varying degrees of lung hypoplasia.23 Left-sided involvement has also been described.24 Diagnosis may be made incidentally in older children or adults, but presentation can occur in infancy, and then is associated with significant mortality related to associated cardiovascular anomalies.25
Horseshoe lung is a malformation characterized by fusion of the right and left lungs posterior to the heart but anterior to the esophagus. In most cases a segment of the right lower lobe extends into the left pleural cavity. There is associated hypoplasia of the right lung and pulmonary artery, dextroposition of the heart, and anomalous drainage of some or all of the right pulmonary veins, all features of scimitar syndrome.22,26 When the left lung provides the abnormal segment, it is hypoplastic and shows similar bronchial and arterial abnormalities. Although horseshoe lung is more often associated with scimitar syndrome, it may have anomalous venous connection, usually supradiaphragmatic, without scimitar syndrome. Horseshoe lung in association with scimitar syndrome may include both severe infantile presentations and milder disease in older children and adults.22,27
Crossover lung segment is a related anomaly. It consists of extension of part of one lung via a similar posterior cardiac/anterior esophageal pathway into the opposite hemithorax, but without parenchymal fusion.22,28
There are no consistent histologic findings in the affected lung or segments in scimitar syndrome, horseshoe lung, or crossover lung segment. Some cases have shown rhabdomyomatous dysplasia (Fig. 6–3) and other dysgenetic changes, but more often there are only secondary changes of acute and chronic infection.
Others
Other positional abnormalities of the lung have been described, largely in association with major congenital abnormalities. Herniation of the lung into the cervical region through an intercostal space, or through the diaphragm, has been described in iniencephaly; similar cervical and diaphragmatic herniations have been seen in Klippel-Feil anomaly; cervical herniation has also been described in the cri-du-chat syndrome. A single apparently normal fetus with intercostal herniation, “bagpipe lung,” has also been described.29
Intrinsic Airway Abnormalities
Tracheal Stenosis
Congenital tracheal stenosis is related to either intrinsic narrowing or extrinsic compression. Acquired stenosis is related to obstructing lesions and scar formation following endotracheal intubation and suction injury. Congenital tracheal stenosis usually presents in infancy with stridor and respiratory distress; older children may present with recurrent pneumonia.30 Intrinsic stenosis may be diffuse (30%), funnel-shaped (20%), or segmental (50%). In diffuse stenosis there is either posterior fusion of tracheal cartilage with the formation of complete cartilaginous rings along the length of the trachea, or a cartilaginous sleeve without regular ring formation. When there are complete cartilaginous rings, the trachea, but not the bronchi, is narrowed. When there is cartilaginous sleeve formation, the bronchi may also be involved. Cartilaginous sleeve formation has been reported in association with various forms of craniosynostosis including Crouzon’s disease and Apert and Pfeiffer syndromes.31 In funnel-shaped stenosis, the upper trachea is normal, but there is a gradual reduction in luminal diameter related to partial or complete loss of the pars membranacea. This form is virtually always associated with other developmental anomalies and has been seen with unilateral pulmonary agenesis.32 It is often associated with left pulmonary artery sling with abnormal origin of the left from the right pulmonary artery and abnormal left pulmonary arterial course posterior to the right main bronchus and trachea and anterior to the esophagus to the left pulmonary hilum. In the stenotic lower trachea, there is complete cartilaginous ring formation, despite the extrinsic compression.11,33 Segmental stenosis is more common than the other forms and may be related to local narrowing by complete cartilaginous rings, an hourglass-like region of narrowing, or focal muscular hypertrophy.34,35 Any part of the trachea may be affected. Extrapulmonary developmental abnormalities are frequently seen with segmental stenosis.
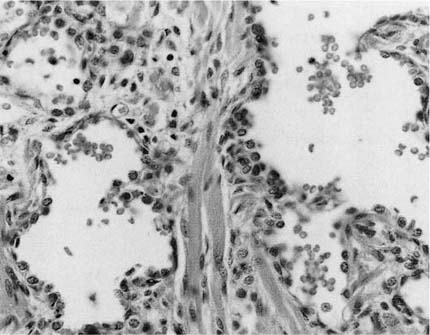
FIGURE 6–3 In rhabdomyomatous dysplasia striated muscle fibers are prominent within the interstitium of abnormally developed lung, as in this hypoplastic and dysgenetic right lung from an infant with scimitar syndrome. (Courtesy of Dr. C. Zuppan.)
Extrinsic stenosis is most often related to vascular compression. Such compression may be due to a vascular ring with double aortic arch, right aortic arch with left ligamentum arteriosum, retroesophageal right subclavian artery, leftward origin of the innominate artery, anomalous left carotid artery, left pulmonary artery sling, or right aortic arch with aberrant left subclavian artery.11
Bronchial Stenosis
Isolated congenital stenosis of a main-stem bronchus has been seen associated with anomalous cartilage segmentation, and reactive changes of atelectasis and squamous metaplasia.36 This is a rare lesion compared with extrinsic partial obstruction or segmental atresia, both of which are associated with lobar overexpansion. Bronchial stenosis is more often a component of tracheal stenosis, particularly the complete cartilaginous sleeve and funnel types. Like tracheal stenosis, extrinsic compression is more common than intrinsic narrowing and is most frequently associated with congenital heart disease with enlarged pulmonary arteries or an enlarged left atrium.37
Tracheo/Bronchomalacia and Tracheal Dyskinesia
Tracheo- and bronchomalacia may be congenital, but are more often acquired, related to damage to the trachea and bronchi from prolonged intubation and aggressive suctioning.38 In both congenital and acquired malacia the large airways are more compliant and easily compressed with a tendency to collapse, leading to expiratory flow obstruction. In acquired malacia the tracheal and bronchial cartilage is normal appearing, but the airways are dilated and the cartilage more widely separated. Congenital malacia is more likely to be associated with deficient and abnormal cartilage development.
Segmental bronchomalacia has been most commonly reported in the left main-stem bronchus, but has also been described in intrapulmonary locations.39,40 It has been reported in association with other anomalies and various skeletal dysplasias.11,40,41 There are also familial examples of bronchomalacia.41,42
Like bronchomalacia, tracheomalacia is a feature of certain chondrodystrophies, including Ellis-van Creveld syndrome, Langer-type mesomelic dwarfism, and diastrophic dwarfism, although details of the cartilage histology have not been reported. It has also been associated with multiplex congenita or Larsen’s syndrome and Hallermann-Streiff syndrome.43,44
In contrast to tracheomalacia, in which the cartilaginous portion of the trachea is weakened and prone to collapse, tracheal dyskinesia is related to dysfunction of the posterior membranous trachea. It may be isolated, but is more often associated with esophageal atresia, tracheoesophageal fistula, and laryngeal cleft, suggesting a developmental origin.45 Both tracheomalacia and tracheal dyskinesia generally show improvement or resolution of symptoms with increasing age.
Congenital Tracheobronchomegaly and Congenital Bronchiectasis
Bronchiectasis is most often an acquired abnormality due to chronic airway infection, sometimes in the setting of mechanical obstruction or immunodeficiency, and these acquired airway abnormalities are discussed elsewhere. Congenital enlargement of the trachea and bronchi may be seen in tracheobronchomegaly (Mounier-Kuhn’s syndrome) and in Marfan syndrome.46–49 Tracheobronchomegaly implies a tracheal and/or main-stem bronchial diameter at least 3 standard deviations above normal.47,50 Tracheobronchomegaly has been seen in association with cutis laxa and Ehlers-Danlos syndrome, suggesting that enlargement of the trachea and bronchi may be a component of various defects in connective tissue formation.47,48 Congenital bronchiectasis (Williams-Campbell syndrome) has also been described.51–56 In all cases there is uniform and symmetric cartilage absence or deficiency beyond the segmental bronchi. Presentation is early, generally within the first year of life, and symptoms include cough, wheezing, and recurrent fever. The generalized deficiency in bronchial cartilage favors the development of bronchiectasis. Familial cases have been seen, suggesting that genetic factors may be important.54,56,57 There are also examples of more focal deficiency in bronchial cartilage.
Congenital Respiratory Tract–Biliary Fistula
Tracheo- and bronchobiliary fistulas are rare anomalies in which a tract connects either the lower trachea or one of the main bronchi near the carina with the biliary tree.58,59 Clinically, patients have bile-stained sputum and sometimes recurrent aspiration pneumonia. Abnormalities of the respiratory tract, particularly esophageal atresia with tracheoesophageal fistula, and of the biliary tree, including biliary atresia, are frequent associations.59–61
Pulmonary Parenchymal Maldevelopment
Acinar Dysplasia/Aplasia
Diffuse pulmonary parenchymal maldevelopment is a rare occurrence. The most severe form of this is the condition initially described as “acinar dysplasia” and “acinar aplasia.”62,63 This condition was later misclassified by Stocker64 as “cystic adenomatoid malformation type 0,” a misnomer as this is not a focal maldevelopment but a diffuse process. Although all reported examples of acinar dysplasia/aplasia have been in infant girls, it has been seen in infant boys as well. These infants, born at term, have the immediate onset of severe respiratory distress and cannot be resuscitated. On examination, the lungs are small and contain essentially only airway structures, bronchi, and sometimes bronchioles, embedded in abundant loose mesenchyme (Fig. 6–4). There is very little or no evidence of acinar development with no alveoli evident. This form of maldevelopment likely results from an abnormal interaction between lung mesenchyme and the bronchial bud, resulting in failure of acinar structures to form, an arrest of lung development in the pseudoglandular stage. The airway structures present are generally normally differentiated, but there is failure of subsequent phases of development. Although this condition is extremely rare, familial cases have been reported, suggesting a genetic basis for this abnormality.65 Studies have show a decrease in both transforming growth factor-β (TGF-β) and type I and type II receptors for TGF-β in the bronchial epithelium and muscularis with acinar dysplasia, suggesting that a reduction in the signaling action of TGF-β during lung development may result in this severe form of pulmonary maldevelopment, although other molecular abnormalities have also been reported.66–68
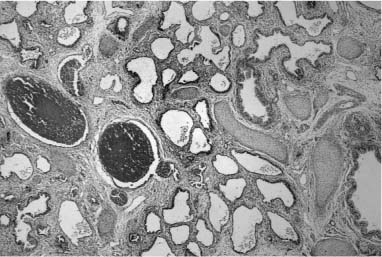
FIGURE 6–4 In acinar dysplasia/aplasia the lung is small and abnormally developed, with only airway development and no acinar structures, as in this small and maldeveloped lung from a 34-week-gestation girl, who had immediate respiratory distress at birth and was difficult to ventilate and oxygenate. She survived less than 1 day. Note the abnormally located cartilage plates in the left lower portion of the photograph. (Courtesy of Dr. K. Meagher-Villemure.)
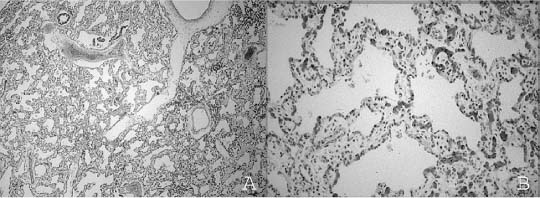
FIGURE 6–5 In congenital alveolar dysplasia. (A) The lung is maldeveloped, showing an arrest in the saccular stage with simple and poorly subdivided saccular spaces forming the lobular parenchyma. (B) There are often widened airspace walls that have a reduction in capillary number, loose mesenchyme, and minimal alveolar epithelial hyperplasia, as seen in the lungs of this infant born at 38 weeks’ gestation and surviving for 1 month, unable to be weaned from ventilatory support. (Courtesy of Dr. G. Jevon.)
Congenital Alveolar Dysplasia
There are somewhat more numerous, but much less well characterized, cases in which arrest of lung development appears to occur at a later stage in the canalicular or early saccular period (Fig. 6–5). Affected infants usually present early in the neonatal period with respiratory and sometimes cardiac insufficiency, and some have pulmonary hypertension as well. They may survive for some time postnatally with maximal ventilatory support.69 Such conditions may be difficult to separate from acquired parenchymal abnormalities related to ventilatory support. Although this condition has not yet been well described, it has been suggested that it may be identical with that initially described as “congenital alveolar dysplasia” by MacMahon.70 Cases reported as “alveolar capillary dysplasia without vein mis-alignment” may also belong in this spectrum.71
Alveolar Capillary Dysplasia with Misalignment of Pulmonary Veins
The developmental malformation known as alveolar capillary dysplasia with misalignment of pulmonary veins is a combined pulmonary vascular and parenchymal defect.72–74 This maldevelopment is familial with a pattern of inheritance that suggests an autosomal-recessive disorder.75–78 Affected infants, usually term or near-term, present early in the neonatal period with respiratory distress and have severe persistent pulmonary hypertension on examination.74 They fail to maintain a sustained response to therapeutic interventions, including extracorporeal membrane oxygenation (ECMO) and inhaled nitric oxide, and usually die within a few days to weeks, although longer survivals and later presentations occur.79 Histologically, there is a characteristic constellation of changes that includes malposition of pulmonary veins adjacent to small pulmonary arteries in both their preacinar and intraacinar course, marked medial hyperplasia of small pulmonary artery branches with extension of arterial smooth muscle into quite small intraacinar vessels, and a deficit in alveolar capillaries (Fig. 6–6).74 Interlobular septa have a marked decrease in small veins, but larger veins may be present. There is often maldevelopment of the acinus with reduction in volume and simplification in structure. Pulmonary lymphangiectasis is also present in about one third of cases. A large proportion of affected infants have other extrapulmonary developmental malformations, often minor, but occasionally major.78 Early reports of cases all had diagnosis at postmortem examination, but many are now diagnosed at lung biopsy when an infant with persistent pulmonary hypertension of the newborn (PPHN) fails ECMO and nitric oxide therapy. The primary developmental defect is unknown. It is not understood whether the vein malposition leads to lobular maldevelopment, whether abnormal lobular development produces an approximation of artery and vein, or whether these two processes are related. In the spectrum of cases there is considerable variability in lobular maldevelopment, whereas vein malposition is a consistent finding, suggesting that the vascular maldevelopment may be primary. The variability in parenchymal maldevelopment may account for the variability in age at presentation and length of survival, but no systematic study of this issue has been done.
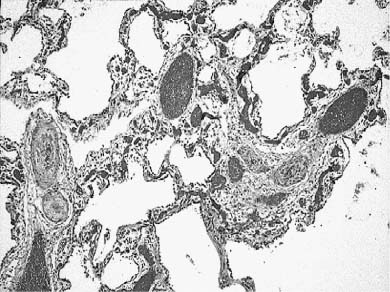
FIGURE 6–6 Alveolar capillary dysplasia with misalignment of the pulmonary veins shows striking hypertensive changes in the small pulmonary arteries adjacent to bronchioles and in their course in the lobular parenchyma. These markedly thickened arteries are accompanied by dilated and congested thin-walled veins, both in their peribronchiolar and intralobular locations. The lobular structure is often simplified, seen here as markedly enlarged airspaces, and the capillary density is reduced in alveolar walls. These changes are particularly striking in the lung of this term male infant who had a late presentation at ~2 weeks of age with pulmonary hypertension. He had failure to thrive but was at home until the onset of a respiratory viral infection at 5 months of age. Diagnosis was made by lung biopsy at 6 months, and he survived to 8 months of age. (Courtesy of Dr. D. Schofield.)
Pulmonary Hypoplasia
Pulmonary hypoplasia is also associated with diffuse maldevelopment of the lung parenchyma that ranges from subtle to extreme, depending on the mechanism of the hypoplasia and in some cases the length of time that mechanism has been active. This maldevelopment was addressed in greater detail in Chapter 2.
Focal Developmental Abnormalities of the Lung (Congenital Lung Malformations)
These lesions are broadly classified as cystic disorders of the lung, although many are not truly cystic. They have been widely recognized for many years, but their origins are only now becoming understood, as information gained through the widespread use of in utero ultrasound has fundamentally changed knowledge in this area. Because of the now nearly uniform ultrasound screening of fetuses, prenatal identification of these malformations is routine, and algorithms for interventional therapy have been developed. Despite the increased recognition of these lesions, definition and classification remain problematic, with different terms applied to similar malformations and the same term applied to disparate conditions. A revised classification has been proposed (Table 6–2) that divides these lesions into four groups: bronchopulmonary malformations, pulmonary hyperplasias, congenital lobar overinflation (emphysema), and other cystic lesions.80
Bronchopulmonary Malformations
Bronchopulmonary malformations include bronchogenic cyst, extralobar sequestration, large cyst–type cystic adenomatoid malformation, and bronchial atresia of various forms both with and without small cyst–type cystic adenomatoid malformation.
Bronchopulmonary malformation |
Bronchogenic cyst (noncommunicating bronchopulmonary foregut malformation) |
Extralobar sequestration |
Without connection to gastrointestinal tract (with/without small cyst–type cystic adenomatoid malformation or rhabdomyomatous dysplasia) |
With connection to gastrointestinal tract (with/without parenchymal malformation as above) |
Cystic adenomatoid malformation, large cyst type (Stocker type 1) |
Isolated |
With systemic arterial/venous connection (hybrid lesion/intralobar sequestration) |
Bronchial atresia |
Isolated |
With systemic arterial/venous connection (intralobar sequestration) |
With connection to the foregut (intralobar sequestration/complex or communicating bronchopulmonary foregut malformation) |
Systemic arterial connection to normal lung |
Cystic adenomatoid malformation, small cyst type (Stocker type 2) |
Isolated |
Associated with bronchial atresia (bronchial obstruction sequence) |
With systemic arterial/venous connection and bronchial atresia (hybrid lesion/intralobar sequestration |
Pulmonary hyperplasia and related lesions |
Laryngeal atresia |
Solid or adenomatoid cystic adenomatoid (Stocker type 3) |
Polyalveolar lobe |
Congenital lobar/sublobar overinflation (congenital lobar/sublobar emphysema) |
Other cystic lesions |
Lymphatic/lymphangiomatous cysts |
Enteric cysts |
Mesothelial cysts |
Simple parenchymal cysts |
Low-grade cystic pleuropulmonary blastoma |
Modified from Langston C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg 2003;12:17–37.
Bronchogenic Cyst
Bronchogenic cyst and extralobar sequestration, although usually considered to be quite separate entities, are best considered together, as their pathogenesis is closely related. Both are generally considered to arise from accessory bronchopulmonary tissue that has lost its normal connection to the developing foregut, or retains a connection to enteric, rather than airway, structures. In the case of bronchogenic cyst this origin is similar to that of other foregut duplication cysts to which they are closely related, and analogous to enteric duplication cysts, as an aberrant bud from the developing foregut.
In general, bronchogenic cysts are solitary unilocular cystic structures with a wall that recapitulates the bronchial wall, a lining of respiratory epithelium overlying fibrovascular tissue and smooth muscle. Hyaline cartilage plates must be present for diagnosis, and bronchial glands are often present as well. The cysts are filled with mucus or watery fluid, or purulent material when infection supervenes. These cystic lesions are most frequently found in the mediastinum just above the tracheal bifurcation and are often attached to the trachea or main-stem bronchi. They occasionally occur within the lung hilum and parenchyma80 (Fig. 6–7), and have been described in a wide variety of locations from the suprasternal region to below the diaphragm, as well as unusual locations such as the retroperitoneum, pleura, neck, base of the tongue, and in subcutaneous tissues.81–89 They occasionally communicate with adjacent structures, particularly the esophagus and stomach.90,91
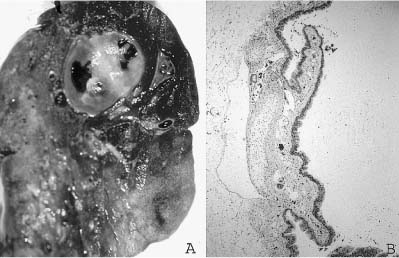
FIGURE 6–7 Bronchogenic cysts. (A) This intrapulmonary unilocular cyst from a 6-month-old boy was filled with clear mucus and has a rather thick smooth wall. (B) Histologically, it is a bronchogenic cyst with a wall that recapitulates the bronchial wall, including cartilage and glands.
Although usually asymptomatic, they may cause airway compression, particularly in young infants with fortuitously located cysts who may develop congenital lobar overinflation in this setting.92–94 The cysts gradually enlarge with increasing body size and age and may reach 10 cm in diameter in older patients. The most common late complication is infection, although hemoptysis and pneumothorax may occur.95,96 When intrapulmonary they do not communicate with the surrounding parenchyma unless there is infection with wall necrosis; however, their presence has a local effect with parenchymal changes that are seen in all examples of in utero intra-pulmonary obstructive lesions.80 Bronchogenic cysts have been reported in association with bronchial atresia and intralobar sequestration.97,98 In the mediastinum they should be distinguished from enteric duplication cysts and neurenteric cysts, and in the lung from abscess.
Extralobar Sequestration
Extralobar sequestration is also thought to result from aberrant bronchopulmonary tissue that develops apart from normal lung tissue. It is defined as an isolated mass of lung tissue with its own pleural investment, separate from the normal lung. Rare bronchogenic cysts have a peripheral rim of parenchymal lung tissue, and many extralobar sequestrations have a large central dilated bronchus or mucocele.80 In such instances these two lesions may be difficult to separate. Although extralobar sequestration is usually located below normally formed lung within the thoracic cavity as a wedge-shaped mass surrounded by pleura,99 it can occur in many of the same locations where bronchogenic cyst has been found. It has been described from the neck to below the diaphragm in the anterior and posterior mediastinum and retroperitoneum, and also has been seen within the diaphragm and pericardium, and as an intraabdominal mass.100–105 In the retroperitoneum it is most frequent in the region of the adrenals, sometimes leading to the erroneous impression of an adrenal tumor on in utero ultrasound examination.106–110
Although extralobar sequestration occasionally communicates with the foregut in any of its locations, typically there is no such connection.111 It generally receives its arterial supply from the descending aorta, but may be supplied from a wide variety of other systemic arteries and occasionally from the pulmonary artery.99,112–114 Venous connection is variable and may be via the pulmonary vein or a systemic vein, particularly the subclavian, hemiazygous, or portal vein.112,114,115 These vascular structures enter the extralobar sequestration via a pedicle at the medial pole. If a bronchus is found within the pedicle, a communication with the foregut should be suspected. The mass of the lesion usually includes a bronchus, which is atretic proximally, and surrounding lung parenchyma (Fig. 6–8). The bronchus is often dilated and may be filled with mucus forming a mucocele, analogous to that seen in bronchial atresia in conventionally located lung parenchyma. The parenchymal structures are maldeveloped, with about half showing microcystic maldevelopment identical to that seen in many examples of conventionally located bronchial atresia.99,100,104,105,116,117 Those both with and without microcystic maldevelopment show parenchymal features of pulmonary hyperplasia identical to that seen in other lesions associated with airway obstruction.80 In this setting of abnormal parenchymal maldevelopment, as in others, there may be rhabdomyomatous dysplasia with striated muscle fibers present within maldeveloped lung tissue.100,117 A few extralobar sequestrations have prominent dilated lymphatic channels within their pleural investment and interstitium.
Extralobar sequestration has been thought to be rare, only found at autopsy with associated developmental abnormalities such as congenital diaphragmatic hernia, and when large producing a mass effect leading to fetal death. These lesions are now identified more frequently on prenatal ultrasound scan in both typical and atypical locations. Those located below the diaphragm have an excellent outcome, but need to be distinguished from other more common lesions in this location.108,118 Intrathoracic lesions are sometimes associated with the development of a large hydrothorax, which may lead to fetal hydrops, polyhydramnios, and fetal death. These large effusions may require thoracoamniotic shunting.119 Fetal intervention is not required in any other circumstance for extralobar sequestration.
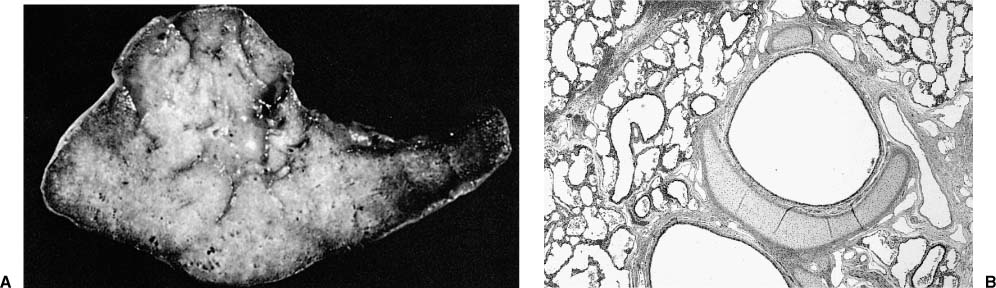
FIGURE 6–8 Extralobar sequestration. (A) The cut surface of this extralobar sequestration shows spongy tissue without evidence of scarring or inflammation. This mass of lung tissue was separate from the normal lung, and located beneath the normal left lower lobe, just above the diaphragm. It received its arterial supply from an intercostal artery. (B) Histologically, extralobar sequestrations contain dilated bronchi and abnormally developed parenchyma that here shows marked airspace enlargement and simplification.
Cystic Adenomatoid Malformation
A heterogeneous group of cystic and noncystic lung lesions was classified into three types by Stocker et al120 in 1977 on the basis of cyst size and microscopic appearance. It is now clear that the least common of these, the adenomatoid or type 3 cystic adenomatoid malformation, is a form of pulmonary hyperplasia, and will be discussed later in the chapter (see Pulmonary Hyperplasia and Related Lesions).121 The two cystic types have been classified as large cyst or type 1 lesions with multiloculated cysts greater than 2 cm in diameter and small cyst or type 2 lesions with more uniform tiny cystic structures usually much less than 2 cm in diameter. The small cyst lesion is generally seen in the setting of parenchymal maldevelopment in extralobar sequestration and bronchial atresia and is discussed with those entities.
The large cyst–type of developmental malformation, termed type 1 cystic adenomatoid malformation by Stocker et al,120 is a distinctive lesion. It is usually single, affecting only one lobe of the lung, but is often multiloculated with occasional smaller cysts that merge with the adjacent lung parenchyma. Although these lesions are typically referred to as cysts, they are in reality cystically dilated structures resembling preacinar airways, with much of their wall having a bronchiolar configuration, a lining of bronchiolar epithelium overlying fibroelastic tissue, and small amounts of smooth muscle, but no glands or cartilage (Fig. 6–9). They are not true cysts and are in communication, albeit distorted, with both proximal airways and the distal lung parenchyma. Some, but by no means all, also contain variable amounts of mucigenic epithelium resembling gastric mucinous epithelium, highlighting the embryologic origin of the lung as a foregut derivative.122 The lung parenchyma distal to the malformation shows maldevelopment similar to that seen in other obstructing lesions. Up to 25% of these lesions have an associated systemic arterial supply; such lesions are more usually seen in the left lower lobe.123–125
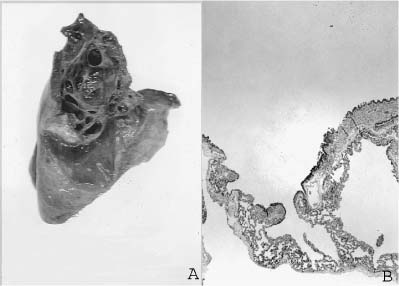
FIGURE 6–9 Large cyst–type cystic adenomatoid malformation. (A) This 6-month-old boy has a large multilocular cystic lesion occupying the apical portion of the right lower lobe. The outlines of the lobe are not distorted by this process. (B) Histologically, the lesion shows the characteristic features of large cyst–type cystic adenomatoid malformation with bronchiolar wall structures making up the “cyst” lining. The characteristic communication with the distal lung parenchyma is evident from the large “cyst.” The distal lung parenchyma is maldeveloped and shows airspace enlargement and simplification.
The large cyst–type cystic adenomatoid malformation is generally thought to present most commonly in infancy or early childhood with respiratory distress due to air trapping, as gradual expansion of the lesion causes compression of adjacent lung tissue and sometimes mediastinal shift. With the widespread use of in utero ultrasound screening, these lesions have been found to have a far more variable presentation and outcome. Most such lesions identified in utero show either marked improvement or spontaneous regression across gestation, and may not require surgical removal.126 Occasionally, they become so large as to impair the development of the normal lung, leading to pulmonary hypoplasia on the basis of a large space-occupying lesion. In this situation there is increased morbidity and mortality even with successful resection when respiratory insufficiency and pulmonary hypertension become apparent. They are rarely associated with the development of fetal hydrops and polyhydramnios and their consequent poor fetal outcome.127 Occasionally, these large cyst lesions may not become symptomatic in childhood and may present in older children or adults either fortuitously or when infection supervenes.128–131
The differential diagnosis for large cyst–type cystic adenomatoid malformation includes low-grade cystic pleuropulmonary blastoma, intraparenchymal lymphangioma, and pneumatocele, all of which may have similar imaging characteristics.132–135 Studies done in the first few days after birth may suggest a more solid lesion, as there is usually delayed clearance of lung fluid from the region.136 Lung fluid is cleared in a biphasic pattern, with the first phase being mechanical clearance from the airways due to thoracic squeeze during delivery, and the second being more prolonged via lymphatic and vascular clearance. The first phase is impaired in lesions without normal airway connections such as extralobar sequestration, bronchial atresia, and large cyst–type cystic adenomatoid malformation.
Although the risk of subsequent neoplasia has been a rationale for early surgery in the setting of large cyst–type cystic adenomatoid malformation, sarcomas reported to arise in this lesion are now thought to be cystic or partially cystic pleuropulmonary blastoma rather than cystic adenomatoid malformation.132,137–139 Pleuropulmonary blastoma had not been identified at the time of most of these reports. Other malignancies have, very rarely, been associated with large cyst–type cystic adenomatoid malformation including lymphoepithelioma-like carcinoma, pulmonary tumorlets, and squamous cell carcinoma, all as isolated examples.140–142 Bronchioloalveolar carcinoma has been reported in a small number of older children and adults with previously unrecognized, unresected, or incompletely resected large cyst–type cystic adenomatoid malformation.143–147 The outcome of such patients has been uniformly good, suggesting that overgrowth of mucigenic epithelium in this circumstance may not be the biologic equivalent of bronchioloalveolar carcinoma.
Isolated Bronchial Atresia
Isolated bronchial atresia is generally considered to be a rare anomaly, particularly in infants and young children, with usual presentation at an older age, either as an incidental finding on chest x-ray, or with a history of recurrent pneumonia or dyspnea.148–151 In this setting, the left upper lobe is the most common site of involvement, with the right upper and lower lobes rarely involved.151 The atretic bronchus may be lobar, segmental, or subsegmental, and is markedly dilated distal to the obstruction and filled with mucus, producing a distinctive nodular lesion on chest x-ray, and a “mucocele” on pathologic examination. Since the advent of widespread in utero ultrasound screening, bronchial atresia has become a more commonly identified lesion in the fetus and neonate.80,152 In distinction to the distribution in older children and adults, most lesions identified in utero are in the lower lobes and are thought to represent intralobar sequestration or cystic adenomatoid malformation on fetal ultrasound screening. The newborns are rarely symptomatic. Later-detected lesions are sometimes initially thought to represent congenital lobar overinflation, or are identified in the setting of a febrile illness.80 The pathology of these lesions is distinctive and similar, regardless of the lobe involved or the age of the patient at surgery.80 When the atretic bronchus is the lobar bronchus, there is a bulge at the hilum marking the underlying mucocele. On dissection of the hilum, a mucocele is always identified, even in the youngest infants, although it may increase in size with increasing age (Fig. 6–10).
Distal bronchi are often dilated, but less prominently, and may also contain abundant mucus. The lobe is usually enlarged, and when a segmental bronchus is involved, poorly developed pseudofissures may outline the affected region on the pleural surface. The parenchyma appears hyperexpanded, and histologically there is airspace enlargement and simplification (Fig. 6–11) and decreased airway and vessel density, as seen in pulmonary hyperplasia and other lesions with airway obstruction.80 In addition to this generalized parenchymal maldevelopment in the affected region, microcystic maldevelopment (Stocker type 2 or small cyst–type cystic adenomatoid malformation) is often focally present. This more focal maldevelopment is seen in ~50% of cases of isolated bronchial atresia, a similar incidence to extralobar sequestration.80,99,100 Other less common and unnamed developmental abnormalities may affect airways and airspaces in occasional cases of isolated bronchial atresia. These include peculiar airway wall widening with increased collagen and proliferated and dilated microvasculature, peculiar cystic lesions, and marked variation in airspace size. Additionally, there may be superimposed chronic inflammation, particularly in older children and adults.80
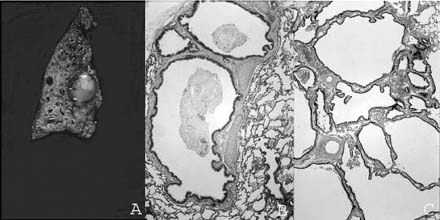
FIGURE 6–10 Bronchial atresia. (A) A large mucocele is evident at the hilum of this maldeveloped left lower lobe. There was no lobar bronchus evident at the hilum. Histologically, there is (B) prominent airway dilatation with contained mucus and parenchymal maldevelopment with both enlarged and simplified airspaces, and (C) microcystic maldevelopment with dilated, tortuous, and numerous bronchiole-like structures separated by variable amounts of simplified parenchyma. The vascular connections, both arterial and venous, were normal. The maldevelopment was noted on in utero ultrasound examination, and lobectomy was done in the neonatal period.
The pathogenesis of bronchial atresia remains unknown. Because the airways both proximal and distal to the atretic region are present, and, although distally dilated, normally distributed, it is generally assumed that bronchial atresia is an acquired abnormality in an initially normally formed airway, rather than a primary developmental abnormality. An analogy to intestinal atresia, known to be caused by in utero vascular events, seems appropriate.153 This would mean that atresia could occur at any time after the large airways are formed, with this process complete by 16 weeks’ gestation.154 Differences in timing of the insult leading to bronchial atresia may account for the range of histologic findings.
Bronchial Atresia with Systemic Vascular Connection (Intralobar Sequestration)
When bronchial atresia occurs in the lower lobes, particularly in the left lower lobe, it is often, but not invariably associated with systemic arterial supply and thus meets the classic definition of “intralobar sequestration.” That is, a developmental malformation of the lung composed of isolated nonfunctioning lung segments having no communication with functional tracheobronchial elements in the surrounding lung and with an abnormal systemic arterial supply and variable venous connection. The abnormal systemic arterial supply is typically from the distal thoracic or proximal abdominal aorta, but may be single or multiple, and may arise from a wide variety of arteries including celiac, splenic, intercostal, subclavian, and coronary arteries, and rarely others.155–159 Venous drainage is usually via the pulmonary vein, but systemic venous drainage can occur. Systemic arterial connection to the lung can be seen both as an isolated finding in otherwise developmentally normal lungs and in association with other developmental malformations including bronchial atresia and large cyst–type cystic adenomatoid malformation. Thus, it is best to consider abnormal systemic arterial supply as a variant of the underlying malformation rather than as a separate primary abnormality.80,123,160–162 This also avoids the widespread, but incorrect, broadened usage of the term sequestration to identify any lesion with systemic arterial connection.
Bronchial atresia with retained systemic arterial connection, when seen in infants and young children, is identical to bronchial atresia without systemic arterial connection in its presentation and histologic features, except for a proportion of cases that involve the posterior basal segment of the left lower lobe.80 In these cases, the atretic bronchus is ectopically located adjacent to the systemic artery after it enters the pleura at the margin of the lung, usually quite near the base; such regions are also often outlined by prominent pseudofissures. Rarely, these ectopically located bronchi are not atretic, but have an abnormal communication with the gastrointestinal tract, usually the esophagus.163 The histologic features are similar to isolated bronchial atresia, except for the identification of a prominent elastic artery entering the pleura in an abnormal location. The artery rapidly anastomoses with the intrinsic pulmonary arterial system and loses its distinctive features as a systemic artery. Pulmonary arteries in the affected region may have prominent arterial smooth muscle and can show progressive changes of pulmonary hypertension, but more often the artery become stenotic as it enters the pleura and the vascular changes are less striking. The pulmonary veins of the affected region are usually moderately dilated related to overcirculation. As with isolated bronchial atresia, a mucocele is present just distal to the atretic bronchus, whether normally or ectopically located. Parenchymal maldevelopment is also similar to that seen in isolated bronchial atresia with airspace enlargement and simplification and with a similar proportion of cases showing microcystic maldevelopment.80,164–166 The presence of microcystic maldevelopment does not alter the prognosis, nor should it change management of these cases. It is best to consider microcystic maldevelopment as part of a malformation sequence, related to the airway obstruction, rather than as a separate process. Secondary changes, including superimposed inflammatory changes, organization of intraluminal material, and organizing pneumonia, may be seen in older children and adults, but are not invariably present.
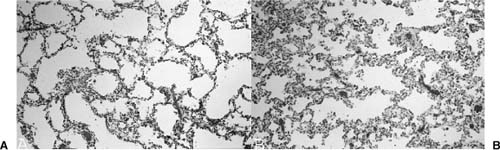
FIGURE 6–11 Postobstructive parenchymal maldevelopment. (A) The lung parenchyma distal to an in utero obstructive lesion shows a typical developmental abnormality that is characteristic of pulmonary hyperplasia with enlarged, simplified, and sometimes thin-walled airspaces. (B) This abnormal development is particularly obvious when there is adjacent normally developed lung parenchyma with more complete alveolarization.
Bronchial atresia with systemic arterial connection, or intralobar sequestration, is clearly a developmental abnormality and has been identified in utero on multiple occasions.124,164,167,168 The previous notion that this particular abnormality could be acquired through the development of systemic collaterals in the face of a chronic inflammatory lesion can no longer be considered to be an appropriate pathogenesis for this lesion. It should be emphasized that collateral systemic circulation to the lung can be encountered in a wide variety of chronic and localized pulmonary diseases including bronchiectasis, tuberculosis, other pulmonary infections, lung tumors, and pulmonary thromboembolism.169,170 Its appearance is quite different from the systemic arterial connection seen in the developmental lesion. In the conditions with acquired systemic collaterals, circulation to the lung is either via anastomoses between bronchial and pulmonary arteries within the lung parenchyma or via transpleural systemic-pulmonary artery anastomoses.171 The most usual origin of systemic collateral circulation is the inferior phrenic arteries, and these anastomoses are usually as a tangled web of vessels at the pleural surface.172
Communicating Bronchopulmonary Foregut Malformation
In addition to the conditions in which normal bronchial connection is absent, such as extralobar sequestration and bronchial atresia in its various forms, there are quite similar malformations in which there is abnormal bronchial connection to the foregut, usually the esophagus, but sometimes the stomach.173–175 Such lesions are termed communicating or complex bronchopulmonary foregut malformations. They are far less common than the more conventional noncommunicating malformations. There is an association between these communicating lesions and the developmental lesions of tracheoesophageal septation that present as the various forms of esophageal atresia/tracheoesophageal fistula. There are rare examples of what has been termed total sequestration or pulmonary ectoplasia175,176 (Fig. 6–12), in which the main-stem bronchus connects with the esophagus rather than the trachea, the lung is usually maldeveloped, and there is a systemic arterial connection. However, these lesions are better classified as communicating or complex bronchopulmonary foregut malformations. The spectrum of lesions with bronchial connection to the esophagus or stomach may be difficult management problems, both before and after surgical correction. Early, the problem is to prevent aspiration of gastric secretions, whereas postoperatively there may be persistent problems with coordinated esophageal function.
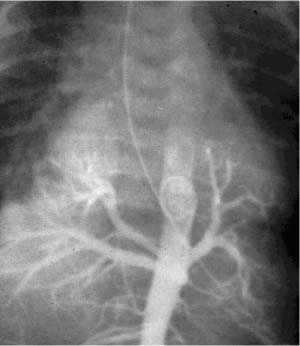
FIGURE 6–12 Pulmonary ectoplasia. This infant has a complex bronchopulmonary foregut malformation strikingly evident on this radiologic examination in which contrast material placed in the esophagus enters the lungs via abnormal bronchial communication on both the right and left sides.
Systemic Arterial Connection to Developmentally Normal Lung
In Pryce’s177 classification of intralobar sequestration, systemic arterial supply to otherwise normal lung was termed a type I sequestration, and is still considered by some to be a form of sequestration. Although this condition is sometimes associated with other cardiovascular developmental abnormalities, in most cases it is asymptomatic and discovered only incidentally. Occasionally in infancy such systemic arterial connection may act as a systemic-pulmonary shunt with associated congestive failure.123 In adults, hemoptysis is a common presenting symptom; massive hemoptysis is rare, but has been reported.178,179 The usual origin of the anomalous systemic artery is from the distal thoracic aorta generally to normal basal segments of the left lower lobe; pulmonary venous drainage is usually normal.180,181 When there is upper lobe involvement, there is usually associated congenital heart disease.182 Histologically, the lung parenchyma appears normal; however, hypertensive vascular changes can occur and may become more severe with increasing age. Lung lobes with systemic arterial supply were formerly removed surgically.123 These lesions are no longer treated by resection, but rather by various forms of embolization and ligation of the anomalous artery.183–185 Untreated, there may be progressive congestive heart failure and severe hemoptysis.186
Anomalous systemic arteries to normal lung and those to lobes containing a variety of developmental abnormalities likely represent the persistence of primitive postbranchial arteries that supply the lung bud prior to development of the main pulmonary arteries.182 These arteries usually extend through the inferior pulmonary ligament but are structurally different from bronchial arteries that may also be present in this location. Structurally, the anomalous systemic arteries are elastic arteries, like the aorta and pulmonary arteries, rather than muscular arteries, like the bronchial arteries. There is often luminal stenosis near their anastomosis with pulmonary arteries, protecting the pulmonary circulation from the elevated systemic blood pressure. It is important to remember that these retained embryonic vessels can be present both as an isolated abnormality and in association with developmental abnormalities within the lung parenchyma, particularly in the lower lobes. Their prominent association with focal developmental abnormalities within the lung suggests that such lesions may impair the normal involution of these embryonic arteries, and their more common occurrence in the lower lobes, may relate to the later involution of the lower embryonic arteries.
Microcystic Maldevelopment (Cystic Adenomatoid Malformation, Type 2)
The microcystic pattern of maldevelopment, classified as “type 2 cystic adenomatoid malformation” by Stocker and coworkers120 in 1977, is usually associated with lesions producing airway obstruction rather than being seen in isolation. These focal lesions are composed of more uniform and smaller cysts, generally less than 2 cm in diameter. Histologically, there is regional replacement of the lung parenchyma by microcystic maldevelopment with a somewhat variable pattern generally involving an increase in enlarged and irregular bronchiole-like structures with variable amounts of intervening alveolated parenchyma (Fig. 6–13), although rarely there is no intervening alveolated parenchyma. The alveolated parenchyma always shows the typical pattern of postobstructive parenchyma with enlarged and simplified airspaces.80 Since Stocker et al’s description, this form of maldevelopment has been widely identified in lesions in which there is bronchial obstruction during development.100,107,116–118,164–167 Such lesions have sometimes been termed hybrid lesions.164 Although there may be isolated examples of this pattern of pulmonary maldevelopment in which airway obstruction is not evident, most such lesions are now thought to be related to in utero airway obstruction.80
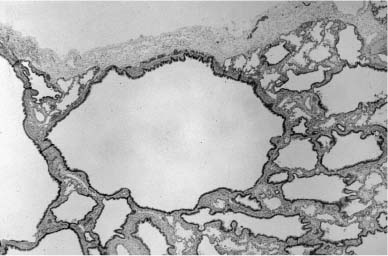
FIGURE 6–13 Microcystic maldevelopment in bronchial atresia. With airway obstruction in utero there is often associated microcystic maldevelopment (Stocker type 2 cystic adenomatoid malformation) with dilated and irregular bronchiole-like structures clustered together with little to no intervening airspaces.
Pulmonary Hyperplasia and Related Lesions
Pulmonary hyperplasia, or excessive growth of the lung parenchyma, is, like pulmonary hypoplasia, a secondary phenomenon.121 During development, it appears to be a response to airway obstruction, and is seen in a wide variety of such situations ranging from laryngeal atresia to more focal lesions. It was identified as “type 3 cystic adenomatoid malformation” in fetuses and stillborn infants with massive enlargement of one or more lung lobes.120 It is this particular component of developmental malformations that can be seen on prenatal ultrasound as brightly echogenic masses that enlarge and often regress. This echogenic appearance, when bilateral and diffuse, is characteristic of laryngeal atresia, but, when unilateral and localized, may be the appearance of a wide variety of developmental abnormalities ranging from bronchial atresia to congenital lobar overinflation.187 This finding suggests that the parenchymal maldevelopment seen in all these conditions has a similar pathogenesis—airway obstruction. The histologic picture varies somewhat, depending on the gestational age of the fetus; however, in all instances it shows a striking increase in airspaces in relation to airways, often with a decrease or absence of interlobular septa, and with abnormally enlarged and often abnormally shaped airspaces (Fig. 6–14). This same picture is seen in the parenchyma distal to focal lesions in the lung, such as bronchogenic cysts and large cyst–type cystic adenomatoid malformation.80
The Lung in Fetal Laryngeal Atresia
It has been known for some time that the lungs in laryngeal atresia, both isolated and as a component of a malformation syndrome, are markedly enlarged and have the histologic picture of pulmonary hyperplasia, as described above. Fraser syndrome, an autosomal-recessive condition characterized by cryptophthalmos, ear anomalies, syndactyly, renal anomalies, and, in boys, cryptorchidism, has laryngeal atresia as a common but not universal feature. Lethal components of this syndrome include both severe renal anomalies and laryngeal atresia. The pulmonary hyperplasia seen in Fraser syndrome and nonsyndromic laryngeal atresia appears to be related to obstruction of the outflow of fetal lung fluid during development.188,189
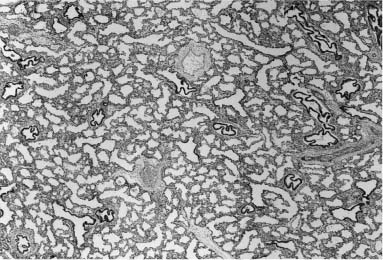
FIGURE 6–14 In all forms of pulmonary hyperplasia, no matter the level of airway obstruction, there is characteristic parenchymal maldevelopment. The histologic appearance varies somewhat with gestational age, for these airspaces do show evidence of maturation in late gestation, although they remain structurally abnormal. This example is from the lung of a 27-week-gestation fetus; compare with Fig. 6–11 from a term newborn.
Adenomatoid or Solid Form of Cystic Adenomatoid Malformation (Cystic Adenomatoid Malformation, Stocker Type 3)
The adenomatoid or solid form of cystic adenomatoid malformation is clearly an example of pulmonary hyperplasia. The more immature appearance of the gas-exchanging parenchyma in early descriptions of this lesion is due to its initial description in preterm fetuses and stillborns.120 The ultrastructural appearance of this lesion is identical to that of normal fetal lungs.190 Although airway obstruction is not a feature of descriptions of these lesions, it is not clear whether it was specifically sought.
Polyalveolar Lobe
Polyalveolar lobe is generally categorized as a variant of congenital lobar overinflation, but it represents a different parenchymal response.191–193 It is a rare condition, but probably underidentified. In polyalveolar lobe, there is marked enlargement of the affected lobe, typically the left upper lobe, with compression of adjacent lobes and mediastinal shift. The alveoli are enlarged, but not as markedly as in congenital lobar overinflation. Morphometric studies have shown a marked increase in alveolar number with a decreased density of bronchioles.194,195 In the polyalveolar regions the airspaces are simplified without prominent subdivisions and resemble those seen distal to obstructing lesions and in other forms of pulmonary hyperplasia. Microcystic malformation (Stocker type 2) has also been associated with polyalveolar lobe.193
Congenital Lobar Overinflation
Pulmonary overinflation in infants and children is a response to airway obstruction, generally of a partial nature. The affected airway may be a lobar, segmental, or subsegmental bronchus (lobar overinflation, and segmental or regional overinflation), and the obstruction may be either congenital or acquired postnatally. Although this lesion is often called congenital lobar emphysema, it is better termed congenital lobar overinflation. Despite its relative frequency in the neonatal population, its identification by in utero ultrasound examination is quite unusual, implying that it has its onset later in gestation after the time of routine ultrasound screening.194–197 With in utero ultrasound the affected region is variously described as “echogenic” and “cystic,” and like other focal regions of pulmonary maldevelopment, has been noted to decrease in size with time.195–197 Affected infants may present in the newborn period with an opacified lobe or sublobar region from which lung fluid is slow to clear.198 Once clearing has occurred, there is gradual enlargement of the lobe, with increasing radiolucency representing overinflation. Affected infants become symptomatic, usually before 6 months of age, as air trapping in the overexpanded region ultimately produces compression of unaffected lung tissue and mediastinal shift. Although lobar regions of overexpansion can be fatal if left untreated, smaller subsegmental regions may not produce symptomatology. Treatment is surgical removal of the affected lobe or segment. The upper lobes, particularly the left upper lobe, are more commonly affected.199
The obstructing lesion may be either extrinsic to the affected airway or intrinsic (within the airway). Intrinsic obstructing lesions are more often partial than complete and include mucus plugs, mucosal flaps or webs, bronchomalacia, other examples of abnormal or defective airway cartilage, and stenosis.11,199–202 Extrinsic obstructions include mass lesions, such as bronchogenic cyst, other localized developmental malformation, and nodal enlargement on an inflammatory basis.203–205 Enlarged or aberrant vessels may also produce extrinsic obstruction. This compression by dilated or aberrant vessels accounts for the occasional association of lobar overinflation with congenital heart disease.201,206–208 Familial cases have also been described.209 Although on gross examination these lobes are often massively enlarged, histologic examination shows only airspace enlargement without any destructive component. Hypoplastic lobes, with prolonged survival, may become markedly overinflated.210,211 This is due to inadequate egress of air from the markedly enlarged airspace via relatively much smaller bronchioles on expiration, leading to progressive overexpansion. When this is more localized, as in the more severely affected lung in congenital diaphragmatic hernia, surgical reduction may be undertaken.
Acquired forms of lobar overinflation have been described as well. These are more frequently seen in intubated infants with injuries to their airways from endotracheal tubes or suction catheters, including granulation tissue polyps leading to partial bronchial obstruction.212 Complete bronchial stenosis with lobar collapse has been described in the same population of infants.213 Segmental overinflation may occur in the presence of a tracheal bronchus.214 Unilateral hyperlucent lung (Swyer-James or Macleod’s syndrome) is an acquired form of pulmonary overinflation related to bronchiolitis obliterans, usually postinfectious in origin.215 Gastroesophageal reflux in infants may rarely be associated with sufficient airway injury to lead to obliterative bronchiolitis, although apneic spells and asthma-like symptoms are more common.216
Other Cystic Lesions
A variety of other developmental cystic lesions may be identified in the lung, although they are far less common than the better-known lesions. These include enteric cysts, identical to foregut cysts seen in other locations; lymphatic cysts, which may be quite large, but collapse with handling; mesothelial cysts; and simple, usually peripheral, parenchymal cysts. Also, cysts that are difficult to classify may occasionally be encountered. Some of these may be examples of known lesions in which distinguishing features are obscured by supervening infection, or by the surgical procedure, and others may show a constellation of previously undescribed features. It is best to classify these as “developmental cystic lesions,” with a descriptive diagnosis, than to place them in a category in which they do not belong.80 Additionally, low-grade cystic pleuropulmonary blastoma has been identified on prenatal ultrasound examination on rare occasions, and sometimes presents in the neonatal period as well. This variant of pleuropulmonary blastoma may have only occasional blastemal or sarcomatous foci. Lesions showing the characteristic morphology of low-grade cystic pleuropulmonary blastoma may occur in neonates and should be extensively examined for these foci.132
Ectopic Tissues Within the Lung
A variety of ectopic tissues have been identified within the lung often in unusual situations. The most common of these is striated muscle, also known as rhabdomyomatous dysplasia,217 in which striated muscle fibers course erratically through the lobular parenchyma. When seen it is often associated with pulmonary maldevelopment, either diffuse with hypoplasia or with a focal developmental abnormality.217–220 It has been described with some frequency in extralobar sequestration,100,118 and in settings with type 2 cystic adenomatoid malformation,120 and may be seen in association with abnormally developed lung in scimitar syndrome.217 Ectopic adrenal cortical tissue and pancreatic tissue have also been rarely described, the latter in association with an intralobar sequestration.221,222 Liver tissue has been described in the lung as well. This is a quite rare occurrence and has always been associated with right diaphragmatic hernia. The liver tissue in this situation does not represent fusion of lung and liver, as it is within the pleural investment of the lung, separate from the liver, and merges with the adjacent lung (Fig. 6–15).223 Neuroglial tissue has also been rarely identified within the lung.224–226 When seen it is often in the setting of anencephaly or craniofacial abnormalities that may permit access of brain tissue to the amniotic space. However, it should be noted, that the vast majority of anencephalic infants do not have pulmonary glial heterotopias. Glial heterotopias are consistently found within peri-airway tissues, rather than within blood vessels and perivascular tissues (Fig. 6–16). Although the etiology remains unclear, its common association with anencephaly has led to the suggestion that at least in these situations it is due to aspiration of fetal brain tissue. Other considerations for which there is less support include embolization of brain tissue, teratoma or hamartoma, and aberrant migration and differentiation of neural crest cells.
FIGURE 6–15 Ectopic liver. This newborn with a right diaphragmatic hernia had ectopic liver evident within the right lower lobe of the lung at autopsy. The lung was small and also has rhabdomyomatous dysplasia, although it appears reasonably well developed, histologically.
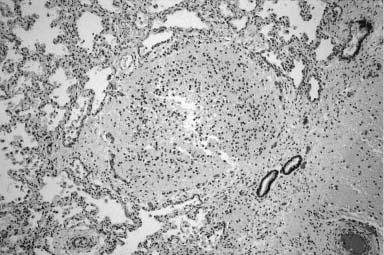
FIGURE 6–16 Neuroglial ectopia. This infant has striking glial implants that completely replace an airway and extend widely in the peri-airway tissues.226
Pulmonary Vascular Disorders
Vascular disorders in the pediatric age group include developmental abnormalities of origin, number, and connection of pulmonary arteries. The spectrum of abnormalities that are often referred to as “sequestration” are discussed above, along with some problems in the nomenclature of these lesions. Changes in the pulmonary vasculature that occur in congenital heart disease are more extensively discussed elsewhere, but those in which the pulmonary vasculature is affected in utero are outlined, as are changes in the vasculature that accompany pulmonary hypoplasia. Both vascular and structural changes in Down syndrome are reviewed. The developmental abnormality known as “alveolar capillary dysplasia with misalignment of pulmonary veins” is addressed above in the section on diffuse developmental abnormalities of the lung, as it combines both vascular and structural abnormalities. The spectrum of persistent pulmonary hypertension of the newborn is discussed. Abnormalities of pulmonary veins and lymphatics that primarily affect infants and children are also included.
Pulmonary Arterial Abnormalities
Arterial anomalies include origin of one or both pulmonary arteries or their major branches from the aorta, its branches, the ductus arteriosus, or the left atrium.11 Origin of the left pulmonary artery from the right and its subsequent course between the esophagus and trachea toward the hilum of the left lung produces a “vascular sling” around the trachea and main bronchi, one of multiple vascular anomalies that can cause respiratory distress via compression of the trachea or by its association with tracheal stenosis due to complete cartilaginous rings (ring-sling complex).227–230
Absent or occult pulmonary artery actually represents interruption of the proximal portion of the vessel, as intrapulmonary branches are usually present and complete.231 Lung development may be affected when the pulmonary artery is absent, with agenesis, hypoplasia, dysgenetic lung with structural maldevelopment, and mesenchymal dysplasia all reported in this circumstance.232–234 A wide variety of abnormalities of the pulmonary vasculature, including pulmonary arterial absence or hypoplasia and/or abnormal pulmonary venous connection, sometimes in association with partial systemic arterial supply, can occur, often in the setting of cardiac malformation. Diffuse hypoplasia of the pulmonary arterial system has been described as a consequence of intrauterine rubella infection.235
Segmental stenosis in the pulmonary arterial system appears in a broad spectrum of disorders.236,237 Stenosis may be single or multiple and peripheral or central. In some instances cardiac anomalies may be present as well. Children with the Alagille syndrome of cholestasis, paucity of intrahepatic bile ducts, and peculiar facies often have peripheral stenotic lesions of the pulmonary arteries.238 Peripheral stenosis has also been described in association with Down syndrome, Ehlers-Danlos syndrome, cutis laxa, Williams syndrome, the LEOPARD syndrome (multiple lentigines, electrocardiographic conduction abnormalities, ocular hypertelorism, pulmonary stenosis, abnormal genitalia, retardation of growth, and deafness), and in association with café-au-lait spots, possibly representing a form of von Recklinghausen’s disease [neurofibromatosis type 1 (NF-1)].11,237,239,240
Arteriovenous Malformations
Arteriovenous malformations (AVMs) occur in the lung as in other organs. They may be single, multiple, or occasionally diffuse. Presentation is usually in older children or young adults, but ~25% of cases occur in early childhood and the newborn period, where presentation is with cyanosis, murmur, or congestive heart failure.241–244 Pulmonary arteriovenous fistulas may develop in a variety of settings and are either acquired on the basis of trauma or following certain palliative cardiovascular procedures, or are primary processes in the lung. They are more common in the lower lobes, and are most commonly seen as part of Osler-Weber-Rendu syndrome, hereditary hemorrhagic telangiectasia, and as such may be familial.245 The arterial supply of the fistula is usually the pulmonary artery but may be systemic. AVMs appear grossly as a focal collection of abnormally dilated vessels of varying size, often with associated thrombosis. The histologic appearance is a tangle of dilated, often congested and occasionally thrombosed vessels with abnormal wall configurations. Complicated vascular malformations involving anastomosis of chest wall vessels to the lung have also been described.246 Rarely the lung may be affected by diffuse telangiectasis, with dilated small vessels providing multiple shunts between the arterial and venous circulations.
Hemangiomatosis and Hemangiomas
Pulmonary capillary hemangiomatosis is another rare vascular lesion of the lung that may present in adolescents and young adults.247–250 It is often associated with pulmonary hypertension. In this lesion there is proliferation of capillary channels, not only in alveolar walls, but also in peribronchial tissues, airway walls, perivascular tissues, and within the walls of larger vessels. This condition has been confused with the striking capillary tortuosity and dilatation seen in some cases of venous obstruction, particularly veno-occlusive disease. It also may be confused with the marked microvascular dilatation and congestion that may be seen in chronically hypoxic infants. In neither of these other conditions is there capillary infiltration of the interstitial tissues of the lung. Pulmonary capillary hemangiomatosis does not occur in infancy, but another disorder, diffuse neonatal hemangiomatosis, does (Fig. 6–17). In diffuse neonatal hemangiomatosis there are widespread capillary hemangiomas of the skin and visceral organs, most commonly the gastrointestinal tract, brain, liver, and lung.251,252 This condition is frequently fatal due to its complications of high-output cardiac failure, gastrointestinal bleeding, hydrocephalus, and consumptive coagulopathy. Therapy with corticosteroids, laser ablation, and interferon-α has had variable but generally unfavorable results.
Localized and multifocal capillary hemangioma may occur in the lungs of infants and young children.253 Cavernous hemangioma is rare, with only a single reported case.254 Localized capillary hemangiomas are seen in infants and young children, similar to the more common hemangiomas of the skin and liver (Fig. 6–18).253 The clinical presentation depends largely on location, with those in the subglottic region and trachea presenting with stridor, wheezing, and retractions, those in endobronchial location presenting with recurrent pneumonia, and those in the lung parenchyma presenting with respiratory distress or being asymptomatic and fortuitously discovered.253 Multifocal capillary hemangioma is identified in only a single report.253 This school-age child presented with shortness of breath, cyanosis, and clubbing; multiple nodules were identified in one lung. Although multiple, the lesions had the typical appearance of capillary hemangioma with small regular, often congested, vascular spaces lined by plump to flattened endothelium. Treatment is generally surgical, although steroids and interferon-α have been used, and except for the multifocal lesion, results are generally good.253
Developmental Pulmonary Vascular Abnormalities in Congenital Heart Disease
Pulmonary hypertension, its pathophysiology, pathologic features, and causes, is discussed later (see Persistent Pulmonary Hypertension of the Newborn). However, it is important to note here that the pulmonary vasculature is structurally different in the fetus, neonate, and young infant from that of older children and adults, making the grading of lesions associated with pulmonary hypertension problematic in the very young.255–259 These anatomic differences led to suggestions for modification of the Heath-Edwards grading system of hypertensive pulmonary vascular changes for infants and young children. This modification required evaluation of the diameter and thickness of artery walls, the peripheral extension of muscle along the vascular tree, and the alveolar/arterial ratio.255,258,260 It never achieved widespread application and has largely fallen into disuse, as it has not been widely reproducible in clinical practice, and its predictive value is quite inconsistent.256,261–263 It has been suggested that the small peripheral biopsies used for this grading may miss irregularly distributed lesions,264 although it is more widely believed that such biopsies are representative of the vascular status of the entire lung.265,266 In any case, the measurements made at echocardiography and cardiac catheterization have been found to be more reliable and less invasive modalities than evaluation of the vasculature at lung biopsy. It is unusual to see high-grade lesions in infants or young children, and if they are present, it is virtually certain that there is some additional factor beyond the cardiac abnormality producing compromise of the pulmonary vasculature.
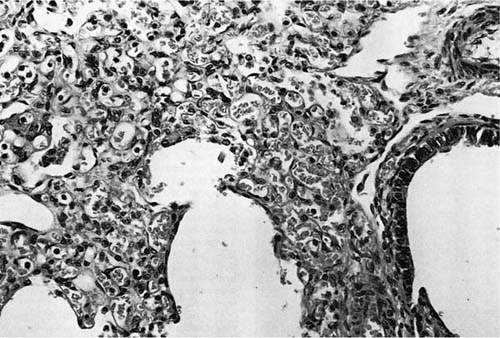
FIGURE 6–17 Diffuse neonatal hemangiomatosis. The lung may be involved in diffuse neonatal hemangiomatosis and shows thin-walled vessels that ramify in the connective tissue of bronchovascular bundles and adjacent alveolar wall. This infant developed heart failure. (Courtesy of Dr. B. Dahms.)
Certain forms of congenital heart disease are associated with pulmonary vascular changes that have their genesis in abnormal intrauterine pulmonary vascular development. When pulmonary blood flow in utero is entirely ductus dependent, as in pulmonary atresia without significant systemic collateral circulation, in critical pulmonary stenosis, and in tricuspid atresia without intraventricular septal defect, prenatal development of the pulmonary vasculature is abnormal.267 In these situations there is a decrease in number, size, and muscularization of both pre- and intraacinar arteries. The preacinar arteries appear dilated and quite thin-walled in their normal location adjacent to small airways, whereas there is a decreased density of intraacinar arteries. When these conditions are associated with significant systemic collateral circulation, advanced vascular disease may develop rapidly.
Developmental cardiac malformations producing significant obstruction to left ventricular outflow in utero, including critical aortic stenosis, aortic atresia, and hypoplastic left heart syndrome, have entirely ductus-dependent systemic blood flow with cardiac output exclusively from the right ventricle. In this situation there is also abnormal intrauterine development of the pulmonary vasculature related to increased pulmonary arterial flow or increased pulmonary arterial pressure.268 The pulmonary vasculature shows an increase in the number of intraacinar arteries, and increased muscularization of pre- and intraacinar arteries and veins with peripheral extension of pulmonary arterial smooth muscle along the arterial tree. In such cases small pulmonary arteries may be hyperreactive and may not dilate or remodel appropriately postnatally. In other types of congenital heart disease in which there is increased pulmonary arterial flow or pressure, including transposition of the great arteries and ventricular septal defect, there may be abnormal postnatal remodeling of the pulmonary vasculature leading to the rapid evolution of obstructive pulmonary vascular disease.269
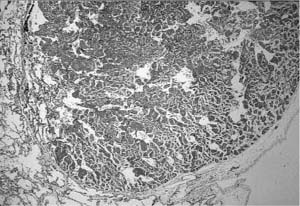
FIGURE 6–18 Capillary hemangioma. Localized capillary hemangiomas of the lung are unusual lesions. This subpleural capillary hemangioma was an incidental finding in the lung of an 8-week-old girl with congenital heart disease. No similar lesion was identified on the skin or in any other organ.
The Pulmonary Vasculature and Lung Structure in Down Syndrome
It is well known that individuals with Down syndrome (trisomy 21 and related chromosomal disorders) who also have congenital cardiac abnormalities have earlier and more severe pulmonary hypertensive changes than chromosomally normal children with similar cardiac defects.270 The structural basis for this is the abnormal postnatal lung development with deficient alveolarization that has been shown to occur in Down syndrome.271 Lung growth and development are abnormal in Down syndrome, with inadequate alveolarization of terminal lung units, widened alveolar ducts, and larger than normal alveoli, whether or not there are associated cardiac defects (Fig. 6–19).272 This growth deficiency results in a reduction in the pulmonary capillary bed, reducing the pulmonary vascular cross-sectional area, exaggerating the effects of intracardiac shunts on pulmonary arterial pressure. Infants and children with Down syndrome with and without cardiac abnormalities may also show retention of the normal fetal double capillary alveolar wall pattern.272 This may also be seen in some non-Down children with cardiac anomalies. The deficient postnatal alveolarization results in air-space enlargement and marked dilatation of subpleural alveoli evident in many children with Down syndrome and is similar to that seen in other children with deficient postnatal alveolarization or with pulmonary hypoplasia. Other patterns of altered lung growth have also been described in Down syndrome.273
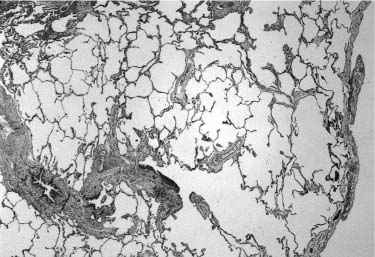
FIGURE 6–19 Down syndrome. The lung shows airspace enlargement that is accentuated in the subpleural regions, but that is evident in all portions of the lobule (compare with bronchiole). Alveolar ducts are also widened. These changes are not striking at birth but become evident in early childhood. This lung biopsy is from a 2-year-old boy with Down syndrome.
Upper airway obstruction is found more frequently in Down syndrome than in chromosomally normal children and may be associated with significant pulmonary hypertension.274,275 This airway obstruction may take various forms and includes laryngomalacia, tracheomalacia, and bronchomalacia, as well as lymphoid hyperplasia, macroglossia, narrow nasopharynx, subglottic stenosis, and tracheal stenosis and commonly multiple sites or modes of obstruction.275 Tracheal bronchus is also a common finding in Down syndrome.275 Congenital tracheal stenosis and tracheal hypoplasia, with an absent or decreased pars membranacea, have also been associated with Down syndrome.276,277
Persistent Pulmonary Hypertension of the Newborn
Persistent pulmonary hypertension of the newborn (PPHN), also called “persistent fetal circulation,” is an important cause of morbidity and mortality in the neonate. This syndrome may be produced by a wide variety of disorders associated with persistent postnatal elevation of pulmonary vascular resistance (Table 6–3), but is most often used to refer to the idiopathic form of the syndrome.255,279–281 Affected infants present with cyanosis, respiratory distress, and evidence of right-to-left shunt with elevated pulmonary artery pressure. Shunting is usually at the level of the foramen ovale, which is often dilated, and there may be ductal shunting as well. There are many known etiologies, some treatable and some not, which must be excluded clinically. PPHN is associated with several primary developmental abnormalities of the lung. Alveolar capillary dysplasia with misalignment of pulmonary veins is well known to present in this fashion, as do certain congenital cardiac defects. The possibility of associated cardiac disease should always be evaluated in the neonate with PPHN, as this may be readily correctable. Pulmonary hypoplasia, whether bilateral or unilateral as in the case of congenital diaphragmatic hernia, is associated with PPHN related to reduction in pulmonary vascular cross-sectional area.278,280 It is virtually always associated with an in utero increase in muscularization of small pulmonary arteries and extension of arterial smooth muscle into normally nonmuscularized intraacinar vessels. In other situations this increase in and extension of arterial smooth muscle is a postnatal occurrence in the setting of severe acquired pulmonary disease. This increase in arterial smooth muscle may result in clinically significant pulmonary vasoconstriction, further complicating disorders such as respiratory distress syndrome, meconium aspiration, and severe pneumonia.
Primary developmental deficit in cross-sectional area of the pulmonary vascular bed |
Pulmonary hypoplasia including congenital diaphragmatic hernia |
Alveolar capillary dysplasia with misalignment of pulmonary veins |
Congenital alveolar dysplasia |
Secondary to failure of postnatal decrease in pulmonary vascular resistance with normal prenatal development (failure of adaptation) |
Large unrestricted ventricular septal defect |
Increased blood viscosity |
Severe acquired pulmonary disease of the newborn resulting in hypoxia |
Respiratory distress syndrome |
Pneumonia |
Secondary to excessive prenatal muscularization of the distal pulmonary vasculature |
Idiopathic |
Associated with meconium aspiration |
Associated with cardiac defects leading to increased pulmonary arterial flow in utero |
Hypoplastic left heart syndrome and related disorders |
Modified from Rudolph AM. High pulmonary vascular resistance after birth. 1. Pathophysiologic considerations and etiologic classification. Clin Pediatr 1980;19: 585–590.
In the idiopathic form of PPHN, it is presumed that a variety of unrecognized stresses to the fetus in utero result in similar abnormal pulmonary arterial muscularization and extension of arterial smooth muscle into normally nonmuscularized intraacinar vessels. Several fetal, placental, and maternal conditions that result in decreased fetal perfusion or oxygenation may precipitate these vascular changes, although it is generally assumed that they must be at least moderately severe, and prolonged for several days or recurrent over a period of days to weeks. It has been suggested that various mediators, particularly leukotrienes, may result in pulmonary vasoconstriction in utero.282 This is thought to facilitate increased arterial muscularization. Premature closure of the ductus arteriosus, usually related to the use of nonsteroidal anti-inflammatory agents late in pregnancy, may also result in increased pulmonary arterial muscularization on the basis of increased pulmonary blood flow in utero.283 Otherevents in which an anatomically normal pulmonary vascular bed might become obstructed leading to PPHN include hyperviscosity and in situ thrombosis or embolization.284,285 Microscopically, PPHN of most sorts is characterized by a distinctive alteration of the normal pattern of muscularization of small pulmonary arteries. In the term fetus and neonate, small pulmonary arteries distal to the level of the terminal bronchiole normally lack a complete muscular layer; that is, there are no muscularized intraacinar arteries.280 Extension of arterial smooth muscle into these initially nonmuscularized vessels is a postnatal phenomenon that normally begins after 6 months of age and is not complete until adolescence. In infants with PPHN there is abnormal extension of arterial smooth muscle into small intraacinar vessels. This vascular remodeling is thought to represent the development of new muscle, as opposed to the persistence of fetal muscle or vasoconstriction.269 These muscularized small vessels are much more likely to be severely compromised by pulmonary vasoconstriction because of their relative size related to the amount of muscle present. In addition, there is often associated proliferation of the connective tissue sheaths of these vessels; this is said to be a response to local hypoxia.
Pulmonary thromboemboli, particularly with associated foreign material, are often seen in the lungs of severely ill infants dying in the neonatal period.286 This foreign material is presumed to be tiny fragments of intravascular catheters or other therapeutic materials employed in the treatment of these very ill neonates and is either a nidus for in situ thrombus formation, which then embolizes, or embolizes itself, and becomes a nidus for thrombus formation in its embolic location.286,287 Nonbacterial thrombotic endocarditis may also be the source of multiple pulmonary thromboemboli, which then may produce significant pulmonary vascular compromise.285
Many infants with both primary and secondary forms of PPHN are now treated with ECMO with varying success. The advent of therapy with nitric oxide, a potent pulmonary vasodilator, marks an advance in the treatment of these disorders,288 although those with PPHN related to abnormalities of lung growth and development may not be capable of sufficient response even with optimum therapies.
Pulmonary Vein Anomalies
In infants and children anomalous pulmonary venous connection, pulmonary vein atresia and stenosis, and pulmonary veno-occlusive disease are the most common primary causes of pulmonary vein obstruction. This obstruction, no matter at what level it occurs, consistently produces the histologic picture of congestive vasculopathy with medial hypertrophy of small pulmonary arteries, thickening of vein walls, engorgement of the microcirculation, edema of interlobular septa, dilatation of lymphatics, peripheral extension of pulmonary arterial smooth muscle into intraacinar vessels, and hemosiderin-laden macrophages.289,290
Anomalous pulmonary venous connection may be either total or partial.281 When total, all four main pulmonary veins converge and then connect with the innominate vein, coronary sinus, or right atrium. Alternately, they may connect below the diaphragm with the inferior vena cava or portal vein. With infradiaphragmatic connection, pulmonary vein obstruction is common, and it may be associated with secondary pulmonary lymphangiectasis. Partial anomalous pulmonary venous connection also occurs. One form is that seen as the defining feature of the “scimitar” syndrome, in which a large anomalous pulmonary vein connects one lung, usually the right, with the inferior vena cava.23,25 The broad, curving image of this vein on chest x-ray forms the “scimitar” that gives the condition its name.291 The affected lung also has bronchial abnormalities (hypoplasia or abnormal connection), a systemic arterial supply (complete or partial), may be abnormally lobated, is often small, and may show parenchymal maldevelopment, often with associated rhabdomyomatous dysplasia.23,25,291
Stenosis or atresia of individual pulmonary veins is a rare congenital defect.291–294
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


