Pulmonary Complications of Sickle Cell Disease
INTRODUCTION
Sickle cell disease (SCD) is one of the most common monogenetic diseases in the world. Sickle cell anemia, the most common and most severe form of SCD, occurs in individuals who are homozygous for a single GAG α GTG substitution in the β-globin gene, resulting in the production of hemoglobin S (HbS). Patients with other types of SCD are compound heterozygotes, having one copy of HbS and one copy of another β-globin mutation, such as hemoglobin SC or HbS-β thalassemia.1 The presence of concurrent α-thalassemia, which will reduce the intracellular concentration of hemoglobin, also modulates disease severity and accounts for some of the variability in the clinical presentation of patients with SCD.2 It is estimated that approximately 250,000 children worldwide are born with homozygous sickle cell (HbSS) anemia every year.3 Approximately 0.15% of African Americans are homozygous for SCD, and 8% have sickle cell trait. In sub-Saharan Africa, up to 40% of the population carry sickle cell trait, and up to 1% of children are born with SCD.4
Despite significant improvements in the life expectancy of patients with SCD, estimates of the median age at death range from 42 to 53 years for men and 48 to 58.5 years for women.5,6 Within this context, pulmonary complications of SCD are common and a major threat to the well-being of patients with SCD, accounting for a large proportion of deaths among these patients.5,7–9 According to the Cooperative Study of Sickle Cell Disease (CSSCD), a prospective multicenter study of 3764 patients, over 20% of adults likely had fatal pulmonary complications of SCD.5 Among the 299 patients enrolled in the long-term follow-up study of patients who participated in the Multicenter Study of Hydroxyurea (MSH) in Sickle Cell Anemia, pulmonary disease was the most common cause of mortality, accounting for 28% of all deaths.10 The numbers likely underestimate the importance of pulmonary disease in risk of death, as pulmonary hypertension (PH) was not diagnosed in these cohorts. Indeed, retrospective evaluation of banked plasma samples from the studies using N-terminal probrain natriuretic peptide (NT-proBNP) as a surrogate for the presence of PH suggested that PH was a major risk factor for death in both the CSSCD and MSH cohorts.11,12
When deoxygenated, HbS is much less soluble than normal hemoglobin (HbA).13,14 Deoxygenated HbS polymerizes and aggregates inside sickle erythrocytes as they traverse the microcirculation. Rigid, dense, and sickled cells can become physically entrapped in the microcirculation, a process that is enhanced by inflammation and integrin molecule expression, causing red cell and leukocyte adhesion to endothelium. Mechanistic studies in transgenic mice expressing exclusively human HbS suggests that microvascular occlusion results in episodic interruption in blood flow, ischemia, and reperfusion injury, with secondary inflammatory, thrombotic, and oxidant stress.15–22
In patients, vasoocclusion leads to the frequent episodes of bone pain and acute chest syndrome (ACS) that complicate SCD. Furthermore, the membrane of erythrocytes containing intracellular HbS polymer is constantly exposed to mechanical and oxidant injury as the red cells traverse the microcirculation. Ultimately, cumulative membrane damage shortens red cell life span, so that SCD is characterized by a chronic hemolytic anemia. Intravascular hemolysis releases cell-free hemoglobin into the plasma, which scavenges nitric oxide (NO) and releases red blood cell arginase-1 into the plasma, which, in turn, catabolizes arginine, the substrate for NO synthesis.23–25 Hence, intravascular hemolysis produces a state of endothelial dysfunction and vascular proliferation, along with prooxidant and proinflammatory stress.26–28 In effect, intraerthrocytic HbS polymerization leads to downstream vascular inflammation, hemolysis-related vasculopathy, and ischemia–reperfusion organ injury.
Anemia, per se, should also be considered in the evaluation of the cardiopulmonary effects of SCD, since decreasing oxygen carrying capacity of the blood related to severe anemia can impair cardiopulmonary function. The signs and symptoms induced by anemia depend on the degree of anemia, the rate at which it evolves, the oxygen demands of the patient, and the presence of chronic cardiopulmonary disease. For instance, in resting adults subjected to acute isovolemic anemia, oxygen delivery can be maintained at hemoglobin concentrations as low as 5 g/dL,29 a finding also noted in individuals with chronic severe anemia.30,31 Thus, anemia, even when severe, rarely causes heart failure; when it does, it is likely that the high-output failure is superimposed upon an underlying cardiac abnormality.
From the hemodynamic standpoint, as hemoglobin level decreases (particularly with hemoglobin values <7 g/dL), blood viscosity decreases, cardiac output increases, cardiac filling pressures tend to decrease, and systemic and pulmonary vascular resistances decrease substantially. Such abnormalities are readily reversible with red blood cell transfusions.30–32 Finally, in severe cases, especially in patients with cardiopulmonary disease, anemic hypoxia can also occur as a consequence of reduced ability of blood to participate in gas exchange and oxygen transport. Interestingly, the severity of hemolytic anemia in patients with SCD is strongly correlated with systemic hypoxemia and decreasing arterial hemoglobin oxygen saturation, a finding that is likely related to both increased cardiac output and altered pulmonary vascular perfusion. Alterations in pulmonary vascular perfusion result in impaired ventilation–perfusion matching.33,34
ACUTE CHEST SYNDROME
The ACS in SCD is defined as a lung injury syndrome characterized by a new pulmonary infiltrate that (1) is consistent with alveolar consolidation, rather than atelectasis; (2) involves at least one complete lung segment; and (3) is accompanied by chest pain, fever, tachypnea, wheezing, or cough.35
 EPIDEMIOLOGY
EPIDEMIOLOGY
ACS is the second most common cause of hospitalization in patients with SCD, the leading cause of admission to an intensive care unit, and, together with PH, a leading cause of premature death, accounting for 25% of SCD-related mortality in earlier cohorts.5,8 More recently, increased awareness, the chronic use of hydroxyurea, and the early and aggressive use of transfusion therapy have led to a decrease in ACS-related mortality.36 For example, in a multicenter trial of inhaled NO for vasoocclusive crisis, only 10% of patients in the study developed ACS, and none required mechanical ventilation or died.36
ACS can occur in any of the sickle hemoglobinopathies but it is more common in individuals with HbSS disease. In the CSSCD, a 29% incidence of the ACS was noted in 3751 subjects over a 2-year period, representing an attack rate of 12.8 episodes per 100 patient-years for homozygous HbS disease.5 The incidence is higher in children than in adults (24.5 events vs. 8.8 events per 100 patient-years). As many as half of episodes of ACS occur in association with vasoocclusive pain crises, and a significant proportion of patients will have a painful event within 2 weeks of the diagnosis of ACS.37,38 Ten to twenty percent of patients admitted with an acute vasoocclusive pain crisis develop ACS within the first 3 days of hospitalization. Recent studies suggest a much higher rate of ACS following influenza infection.39–41
Clinical parameters that appear to increase risk for, or are associated with, development of ACS include: young age, active smoking, environmental smoke exposure,42 major surgical procedures, acute rib infarcts, avascular necrosis of the hips, pregnancy, use of narcotics, acute anemic events, nocturnal or daytime hypoxemia, and previous pulmonary events.37,38 In children, a number of studies suggest that asthma is a risk factor for the development of ACS.43–45 Laboratory parameters in steady state associated with an increased risk for the development of ACS include an elevated white blood cell count, higher steady-state level of hemoglobin, and lower steady-state level of fetal hemoglobin.37,38
While a high steady-state level of hemoglobin is a risk factor for ACS, during acute hospitalization for a vasoocclusive crisis, development of the ACS is often preceded by an abrupt drop in hemoglobin (mean decrease of 0.78 g/dL) and increases in markers of hemolysis, for example, lactate dehydrogenase. The platelet count may also fall prior to ACS, and platelet levels lower than 200,000 cells per mL constitute an independent risk factor for the severity of ACS, an increased risk of neurological complications, and the need for mechanical ventilation.
A limited number of studies have addressed the role of candidate gene polymorphisms in patients with ACS. In a small cohort of 134 pediatric patients with SCD, NOS1 AAT repeat polymorphism in intron 13 was associated with increased risk of ACS.46 Similarly, in a study of 173 pediatric patients with SCD, individuals homozygous for a ET-1 T8002 C polymorphism had higher rates of ACS, while those homozygous for a NOS3 T-786 C polymorphism had lower rates.47 Finally, in a cohort of 942 children with SCD, a highly polymorphic (GT)n dinucleotide repeat located in the promoter region of HMOX1, with long repeat lengths linked to decreased activity and inducibility, was evaluated. After adjusting for sex, age, asthma, percentage of fetal hemoglobin, and α-globin gene deletion, children with two shorter alleles had lower rates of hospitalization for ACS compared with children with longer allele lengths.48
 PATHOGENESIS
PATHOGENESIS
The etiology of ACS is multifactorial. Although a specific cause is not identified in a substantial proportion of patients, three major pathogenetic mechanisms are involved, including infection, bone marrow fat embolization, and direct red cell intravascular sequestration, which cause lung injury and infarction (Table 96-1, Fig. 96-1).
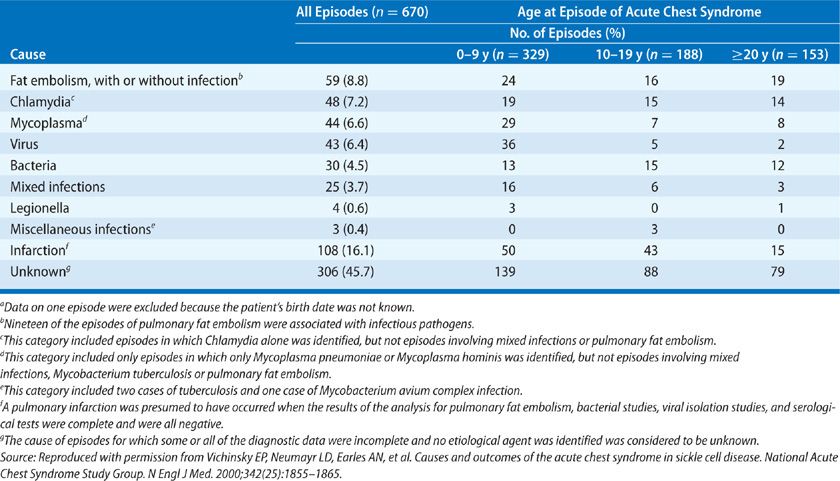
Figure 96-1 Pathogenesis of the acute chest syndrome. Three major mechanisms are associated with the development of ACS: infection, bone marrow fat embolization, and direct red cell intravascular sequestration causing lung injury and infarction. Lung injury results in ventilation–perfusion mismatch and hypoxemia, which leads to increased hemoglobin S polymerization, and erythrocyte vasoocclusion. This worsens bone marrow infarction and pulmonary vasoocclusion to promote a vicious cycle. Fat embolization can be diagnosed by Oil Red O staining of pulmonary alveolar macrophages, revealing the characteristic red lipid inclusions. Common infectious organisms identified in cases of ACS are listed. (Reproduced with permission from Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med. 2008;359(21):2254–2265.)
The most common etiology of ACS in both children and adults is infection by a community-acquired pathogen. It has been proposed that community-acquired respiratory infection induces an excessive inflammatory lung injury response in patients with SCD, as SCD mice that express only human HbS show increased susceptibility to inflammatory triggers (lipopolysaccharide and bacteria) and development of lung injury at lower levels of endotoxin that do not adversely affect wild-type mice.49,50 In addition, more than 80% of adults with SCD report a history of having been admitted to the hospital for “pneumonia.”26
The National ACS Study Group analyzed 671 episodes of ACS in 538 patients with SCD. Respiratory samples obtained from sputum and BAL were analyzed for viral and bacterial infections.35 Among the infectious agents identified most commonly (Table 96-1) were atypical bacteria and viruses, including Chlamydia pneumonia (29%), Mycoplasma pneumonia (20%), Legionella pneumophila (2%), respiratory syncytial virus (10%), parvovirus (4%), rhinovirus (3%), parainfluenza virus (2%), influenza A virus (2%), cytomegalovirus (2%), Epstein–Barr virus (1%), and herpes simplex virus (1%). Community-acquired encapsulated bacteria were rarely isolated, despite the fact that patients with homozygous HbS disease rarely have normal splenic function. Staphylococcus aureus was isolated in 5% of cases and Streptococcus pneumonia in only 4% of cases. Cases of severe ACS related to seasonal influenza have also been described.39–41
Fat embolization syndrome occurs as a complication of vasoocclusive pain crisis involving multiple bones, resulting in bone marrow edema, infarction, and necrosis. Fat embolism was identified by the National ACS Study Group as the cause of ACS in 16% of patients. During fat embolization, bone marrow contents are released into the systemic circulation and are trapped in the pulmonary circulation, producing acute PH, severe lung inflammation, and hypoxemia (Fig. 96-2).51–53 Bone marrow fat released into the bloodstream is also converted to free fatty acids by secretory phospholipase A2, producing direct inflammatory lung injury.54
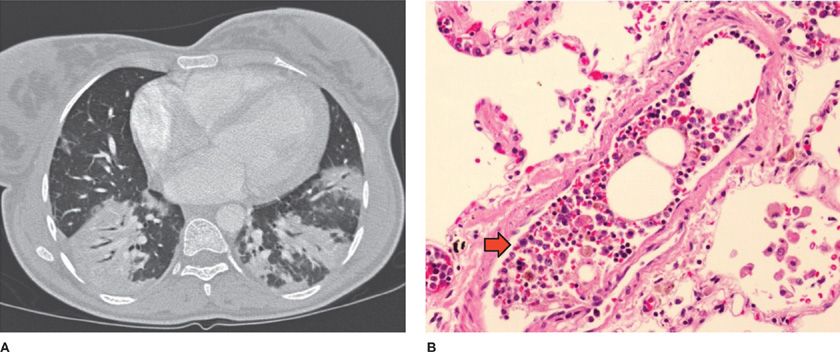
Figure 96-2 Fat embolization in acute chest syndrome. A. Chest CT scan of a patient with acute chest syndrome and fat embolization syndrome. B. Postmortem examination specimen of a patient who died suddenly during an episode of vasoocclusive crisis and acute chest syndrome demonstrating bone marrow elements lodged in the small pulmonary artery (arrow). (Reproduced with permission from Vij R, Machado RF. Pulmonary complications of hemoglobinopathies. Chest. 2010;138(4):973–983.)
In approximately 20% of patients, direct adhesion of sickled cells in the pulmonary vasculature arising during lung infarction or lung vasoocclusion is associated with the development of ACS; a small fraction of patients develop wedge-shaped infarctions, sometimes followed by central cavitation.55 In situ pulmonary arterial thrombosis and cellular occlusion also appear to be common in patients with ACS. In a study of 144 episodes of ACS in 125 consecutive patients evaluated using CT angiography, a 17% prevalence of subsegmental thromboembolism was noted; there was no evidence of peripheral venous thrombosis.56
A potential role for hemolysis-derived plasma-free hemoglobin and its by-products, such as free heme, has been suggested in the pathogenesis of ACS. Administration of lysed red blood cells into the circulation of sickle cell mice has been shown to increase vascular permeability in the lung without impacting permeability in other organs.57 Furthermore, preliminary studies have demonstrated that intravenous administration of heme to sickle cell mice induces severe and lethal acute lung injury.58 In aggregate, these data suggest that plasma-free hemoglobin or heme specifically may directly contribute to lung injury in the context of vasoocclusive crisis and ACS. The relationship between increased intravascular hemolysis and thrombocytopenia (see below) suggests that a possible thrombotic thrombocytopenic purpura–like mechanism may occur in a subset of patients with ACS. In fact, studies have demonstrated that hemoglobin produced during hemolysis may inhibit ADAMTS13 activity.59–61
 CLINICAL FEATURES AND EVALUATION
CLINICAL FEATURES AND EVALUATION
Eighty percent of patients with ACS present with fever and 62% with cough; approximately 40% have chest pain, tachypnea, dyspnea, and abdominal, arm, leg, rib, or sternal pain.35 Some of the clinical features of ACS are age dependent, which likely reflects the different disease etiologies in different age groups. Children have a higher proportion of infectious etiologies in comparison to adults, who have fat embolization as a major cause. Most adult patients present with severe extremity or chest pain and develop ACS 24 to 72 hours later. Reactive airway disease is observed in 13% of cases and is much more common in children.35
ACS is associated with signs of systemic inflammation. Mean peak temperature is 38.9°C and mean white blood cell count is 23,000 cells per mL. In addition, a drop in hemoglobin level (mean decrease of 0.78 g/dL from steady-state level) and an increase in markers of hemolysis are noted.35 Thrombocytopenia may also occur; platelet counts less than 200,000 cells per mL appear to be a marker of more severe ACS and are associated with increased risk of neurological complications and need for mechanical ventilation.35 Secretory phospholipase A2 levels, that are elevated early in the course of ACS, even before development of radiographic changes, have been used to predict onset of the syndrome.62
Some patients manifest evidence of systemic fat embolization. In these cases, ACS is part of the spectrum of the systemic fat emboli syndrome, evident as acute multiorgan system failure (MOSF) (Chapter 142). Patients with MOSF experience acute hypoxic respiratory failure, acute cor pulmonale, renal and hepatic dysfunction, alterations in mental status, seizures, thrombocytopenia, and coagulopathy.63,64
The diagnosis of pulmonary fat embolization syndrome is based on identification of Oil Red O–positive lipid accumulations within alveolar macrophages, as the clinical manifestations can be indistinguishable from other causes of ACS (Fig. 96-2). Bronchoscopy has been used as the diagnostic modality of choice for the diagnosis of pulmonary fat embolization syndrome. Interestingly, a study comparing the diagnostic utility of induced sputum sampling of alveolar macrophages with specimens obtained from bronchoalveolar lavage found a significant, albeit modest, correlation between the two (R = 0.65).65 Patients with ACS who have lipid-laden macrophages detected in induced sputum have significantly more extrathoracic pain than those without evidence of fat emboli, more neurological symptoms, a lower platelet count, and higher serum transaminase levels.65
The mean length of hospitalization for ACS is 10.5 days, compared with 3 to 4 days for uncomplicated vasoocclusive painful crisis. Large registry studies suggest that 13% of patients require mechanical ventilation. The overall mortality is 3% for all patients and 9% for adults, however, as noted previously, these rates may be lower in experienced medical centers employing more aggressive and earlier transfusion therapy.
Risk factors for mechanical ventilation and poor outcome include a platelet count less than 200,000 per μL (likely indicative of the fat emboli syndrome), a greater number of lobes involved on the chest radiograph, and a self-reported or medical record–based history of cardiac disease.
Acute cor pulmonale can also complicate the course of ACS. In a study of 84 consecutive patients hospitalized with ACS, 13% manifested right heart failure. This subgroup had significant elevations in B-type natriuretic peptide and troponin I levels and the highest risk for needing mechanical ventilation and death.66 PH and right heart dysfunction appear to represent major comorbidities in ACS; right heart failure should be considered in those patients presenting with shock or severe hypoxia.
 TREATMENT
TREATMENT
Since the triggers and risk factors for ACS are well known, preventive strategies in the outpatient setting, clinical surveillance, and aggressive and early therapy are likely to improve prognosis.
Patients with multiple episodes of vasoocclusive crises or a previous history of ACS should be treated with hydroxyurea in the outpatient setting, since its use has been shown to reduce the risk of developing ACS by approximately 50%.67,68 A chronic transfusion regimen is also effective in reducing the incidence of ACS,69 as is preoperative blood transfusion in patients undergoing surgical procedures.70
In acutely ill patients, specific strategies, such as aggressive pain management and incentive spirometry, can minimize chest wall splinting, mitigating development of atelectasis and alveolar hypoxia. The use of incentive spirometry has been shown to decrease the incidence of new pulmonary infiltrates in patients admitted with vasoocclusive pain affecting the chest wall.55
In one small study evaluating the efficacy of prophylactic blood transfusions in patients with serum phospholipase A2 elevations during vasoocclusive crisis,71 transfusion eliminated ACS in the study cohort. Larger confirmatory trials are indicated.
We recommend use of empiric antimicrobial therapy in all patients with ACS, given the high prevalence of infectious etiologies. Coverage should include agents effective against atypical bacteria and encapsulated organisms. Important to consider are alternative organisms, such as methicillin-resistant Staphylococcus aureus (MRSA) and influenza viruses, especially in patients who do not respond to therapy or who develop ACS during influenza season.
Although not demonstrated in randomized trials, blood transfusion remains the mainstay of ACS therapy and is considered standard of care. Acute red cell transfusion increases PaO2 and hemoglobin oxygen saturation and may rapidly resolve the pulmonary event.35,72 In the DeNOVO trial,36 no episodes of ventilator-dependent respiratory failure and no deaths were noted in 30 patients with ACS receiving the current practice of early transfusion therapy. The improved outcomes compare favorably with historical data from the National ACS Study Group.
To avoid the adverse effects of increased blood viscosity and the consequent risk of vasoocclusion, the hemoglobin level should not be raised higher than 11 g/dL with simple transfusion therapy. The National Acute Chest Syndrome Study Group found no significant differences in outcomes between patients treated with simple transfusion or red cell exchange (erythrocytapheresis), suggesting that the former approach is preferred as initial therapy.35 Patients with high initial hemoglobin concentrations (≥9 g/dL) or patients with severe or rapidly progressive disease should receive erythrocytapheresis. Most importantly, to decrease the risk of delayed hemolytic transfusion reactions related to alloimmunization against minor red blood cell antigens, all transfused blood should be matched to Rh, C, E, and Kell antigens.
Treatment with corticosteroids has been shown to reduce the severity of pain and length of hospitalization, but this therapy is complicated by a high rate of rebound pain and readmission.73,74 An investigation of the use of a slow, tapering protocol corticosteroids to maintain their beneficial effects while limiting rebound pain and readmission was terminated early because of slow accrual of study patients.75
Noninvasive mechanical ventilation (NIMV) has been studied in the setting of ACS. In a prospective, randomized, open single-center study of 67 adult patients with ACS, use of NIMV improved respiratory rate and gas exchange but failed to significantly reduce the number of patients remaining hypoxemic by day 3 of hospitalization; in addition, use of NIMV was associated with greater patient discomfort.76 In addition, NIMV did not change transfusion rates, pain scores, narcotic dose, or hospital length of stay. In fact, its use prolonged length of stay in the step-down unit.76
The DeNOVO trial explored use of inhaled NO for patients with SCD presenting in vasoocclusive crisis. Despite two prior positive small phase II trials,77,78 the DeNOVO study did not demonstrate an effect of inhaled NO therapy compared with placebo on the duration of pain crisis, narcotic use, pain scores, or development of ACS.36
ASTHMA AND AIRWAY REACTIVITY
Asthma is common among patients with SCD, particularly in children. In a prospective study of 291 African-American children with SCD, 17% received a clinical diagnosis of asthma.43 Interestingly, the reported incidence of airway hyperresponsiveness in children with SCD is much higher, ranging from 40% to 77%, as measured by methacholine, exercise, or cold air challenge tests.79–81
Asthma has been associated with multiple complications of SCD, including ACS. Children with SCD and asthma experience almost twice as many episodes of ACS as their counterparts without asthma, even after matching for age, sex, fetal hemoglobin percentage, and lifetime average hematocrit.43–45,82–85 Several mechanisms have been proposed for this association, including ventilation–perfusion mismatch leading to hypoxemia and sickling, and inflammation causing increased erythrocyte adhesion to vascular endothelium.86 Asthma is an independent risk factor for mortality in children with SCD, conferring a twofold higher risk of death (hazard ratio, 2.36; confidence interval [CI], 1.21–4.62; p = 0.01).87 The reasons for increased mortality remain unclear, but are likely related to the higher risk of vasoocclusive crises and ACS.
Data on asthma in adults with SCD are scarce. The diagnostic and prognostic significance of the disease in adults is less clear than in children. A study of 31 adults with SCD without a clinical diagnosis of asthma showed an average TLC of 73% predicted, FVC of 79% predicted, and FEV1 of 75% predicted.85 Fifteen patients demonstrated bronchial hyperreactivity on methacholine provocation testing; there was evidence of a correlation between bronchial hyperreactivity and previous episodes of ACS. Another cohort study evaluated physician-diagnosed asthma and a history of wheezing in 114 adults with SCD.88 Although self-reported severe and recurrent wheezing was associated with increased rates of pain, ACS, and risk of death, no relationship was found between a physician-established diagnosis of asthma and these complications.
Although there are no controlled trials of asthma therapy in the SCD population, most experts recommend treatment following generally established guidelines for asthmatics in general.89 Systemic corticosteroids must be used with caution, however, as corticosteroids appear to induce vasoocclusive crises and ACS in patients with SCD. In fact, the use of corticosteroids in pediatric patients with SCD has been shown to worsen outcomes.
PULMONARY HYPERTENSION
Among the chronic cardiopulmonary complications of SCD, PH, defined as a mean pulmonary artery pressure (mPAP) ≥25 mm Hg, has emerged as the major threat to the well-being and longevity of patients. A number of studies have added significantly to our understanding of the prevalence, underlying pathogenetic mechanisms, and clinical phenotype of patients with PH and SCD. Several screening cohort studies have focused on the prevalence, risk factors, and mortality rate of patients with SCD and PH diagnosed by right heart catheterization.
 EPIDEMIOLOGY
EPIDEMIOLOGY
Retrospective studies using Doppler echocardiography have demonstrated that 20% to 30% of patients with SCD have an elevated estimated pulmonary artery systolic pressure (PASP) that is two standard deviations above the normal mean value (tricuspid regurgitant jet velocity [TRV] ≥ 2.5 m/s). Approximately 8% to 10% have values three standard deviations above the normal mean (≥3.0 m/s).64,90 These findings have been corroborated in several prospective studies.
In one study using echocardiographic screening, 23% of patients with SCD had borderline or mild elevations in PASP (defined by a TRV ≥2.5–2.9 m/s and corresponding to a PASP of approximately 30–39 mm Hg), and 9% had moderately to severely elevated pressures (defined by TRV ≥3.0 m/s, corresponding to a PASP of approximately 40–45 mm Hg).26 Similar rates were found in other studies.91–94
In a study of patients enrolled in the MSH, plasma NT-proBNP, a prohormone released by right and left ventricular myocardium under pressure stress, was elevated in 30%, suggesting the presence of elevated pulmonary pressures and right heart strain.11 Similarly, measurements of NT-proBNP in banked plasma from patients enrolled in the CSSCD from 1978 to 1988 revealed that 27.6% of adults had elevated levels.12 In both studies, elevated study entry levels of NT-proBNP were independently associated with a higher risk of death in prospective follow-up.
Epidemiological risk factors associated with an elevated TRV include a history of renal or cardiovascular complications, increased systemic systolic blood pressure, abnormalities in markers of hemolytic anemia (anemia; reticulocytosis; and increased lactate dehydrogenase, aspartate aminotransferase, and bilirubin levels), iron overload, cholestatic liver dysfunction (elevations in alkaline phosphatase), renal insufficiency, a history of cutaneous leg ulceration, and, in men, a history of priapism.11,26 These risk factors have also been observed in more recently published studies using right heart catheterization to diagnose PH.93,95,96 Surprisingly, in these studies development of an elevated TRV was not associated with the number of vasoocclusive episodes, markers of inflammation, fetal hemoglobin levels, or platelet counts.11,26 PH determined by right heart catheterization has also been shown not to correlate with episodes of vasoocclusive crisis or ACS, suggesting that this complication arises secondary to hemolytic anemia, rather than recurrent episodes of vasoocclusion. A large screening study of 483 patients with homozygous SS disease conducted in the United States and England reproduced these associations97; patients with elevations in Doppler-estimated PASP had more severe hemolytic anemia and renal insufficiency. These patients also had lower arterial oxygen saturation, higher levels of NT-proBNP, and lower 6-minute walk distances. A high Doppler-estimated pulmonary artery pressure was also independent of rates of vasoocclusive disease or ACS, providing further support to the hypothesis that this complication arises secondary to chronic hemolytic anemia and end-organ dysfunction (renal and liver disease), rather than episodes of ACS and related pulmonary fibrosis.
An elevated estimated PASP by Doppler-echocardiographic screening or right heart catheterization is a significant risk factor for death in patients with SCD. The risk appears to be linearly related to the elevation in PASP.26 In one study, a 14% mortality rate over 2 years was reported in patients with an elevated TRV versus a 3% mortality rate in those with normal TRV.92 Similarly, in another study, the mortality rate over 2.5 years was 25% in patients with an elevated TRV and less than 2% in those with a normal TRV.91 Consistent with these data, in a cohort of 632 patients with SCD from the United States and England, 11.2% had TRV ≥3.0 m/sec and 24.1% had NT-proBNP level ≥160 pg/mL. Of 22 deaths during follow-up, 50% had a TRV ≥3.0 m/sec. At 24 months the cumulative survival was 83% with TRV ≥3.0 m/sec and 98% with TRV <3.0 m/sec. The hazard ratios for death were 11.1 (95% CI 4.1–30.1; p <0.0001) for TRV ≥3.0 m/sec, 4.6 (1.8–11.3; p = 0.001) for NT-proBNP ≥160 pg/mL, and 14.9 (5.5–39.9; p <0.0001) for both TRV ≥3.0 m/sec and NT-proBNP ≥160 pg/mL.98
Several studies have provided new insights into PH in SCD using the gold standard diagnostic test for the disease—right heart catheterization (Table 96-2).



