Pulmonary Complications of Cystic Fibrosis
Kristy Phillips
David R. Jones
Christine L. Lau
Cystic fibrosis (CF) is an autosomal recessive inherited disease caused by mutations in a single gene that encodes the CF transmembrane conductance regulator (CFTR) protein. The major clinical manifestations are due to dysfunction of the CFTR protein and include chronic sinopulmonary disease, impaired exocrine pancreatic function, elevated sweat chloride, and male infertility. Despite major recent advances in understanding the molecular mechanisms and pathophysiology of the disease, CF remains the most common lethal genetic disease in the white population. CF affects approximately 30,000 individuals in the United States and 100,000 worldwide, with an estimated incidence in the U.S. white population of 1 in 3,200.40,42,79 CF may be seen, but less frequently, in nonwhite populations. It occurs in 1 in 9,000 of Hispanic,76 1 in 32,000 of Asian,89 and 1 in 15,000 of African-American births.79 The frequency is approximately equal among males and females.
Dedicated CF centers throughout the world have increased patient survival and promoted research into all aspects of diagnosis and treatment.128 Patients with CF continue to live longer. In the 1950s, children with CF were not expected to live to age 5. The U.S. Cystic Fibrosis Foundation registry tracks health information on 24,000 patients cared for at Cystic Fibrosis Foundation–accredited care centers since 1969.40 In 2005, the median predicted survival was 36.5 years, compared with 32 years in 2000 and 28 years in 1990. In 2005, 43% of people with CF in the United States were >18 years of age, compared with 33% in 1990. Rosenfeld and associates168 reported a gender difference in mortality, with a median age for survival in 1992 of 28.4 years in males and 25.3 years in females. The authors analyzed multiple factors, such as pulmonary function and nutritional status, and found no explanation for the gender difference in mortality. Respiratory failure accounts for 80% of deaths in CF patients in the United States.40
The CF gene is located on the long arm of chromosome 7. Mutations lead to alterations in CFTR protein function. The structure of CFTR was first described by Riordan and associates162 in 1989. The CFTR structure includes two domains that cross the cell membrane and serve as chloride channels. Binding of phosphorylating nucleotides, including adenosine monophosphate, to intracellular domains of the protein is essential for CFTR function.36,197 Using monoclonal antibodies against CFTR, Kartner and coworkers97 confirmed the distribution of CFTR in the epithelial cells of pancreatic ducts, reabsorptive ducts of sweat glands, intestinal crypts, and the submucosal glands of respiratory epithelia. The epithelial tissues of CF patients are impermeable to the passage of chloride ions, which normally are transported through channels in the apical cell membranes. The absence of chloride absorption in the sweat tubules results in the retention of excessive amounts of chloride ions as well as sodium ions and water in the sweat. In the respiratory epithelia, chloride secretion is deficient, and the epithelial cells absorb more sodium than normal. Water passively follows the chloride and sodium ions into the cells, with dehydration of the mucous secretions.153 The exact mechanism by which CFTR protein dysfunction leads to the CF phenotype is unknown. The effect of CFTR protein dysfunction on airway epithelial and glandular cells results in a decrease in the depth of the airway surface liquid layer in the lungs, an increase in the viscosity of airway secretions, and decreased ability to clear bacterial infection.130 In other epithelial and glandular tissues, such as the pancreas and small intestine, dehydrated secretions lead to ductal obstruction, which impairs normal glandular function.
Clinical Manifestations and Diagnosis
Prior to 1989, the diagnosis of CF was confirmed by evidence of an elevated sweat chloride concentration in patients with typical pulmonary or pancreatic insufficiency. Currently, genetic testing can identify patients with unusual variants of the disease who may have normal sweat chloride levels. Newborn screening can identify patients prior to the development of clinical disease.214 However, CF continues to be primarily a clinical diagnosis. Such a diagnosis requires the presence of clinical features as well as laboratory evidence of CFTR protein dysfunction.
A consensus statement on the diagnosis of CF summarizes key aspects of the phenotype.169 These include:
Chronic sinopulmonary disease as manifested by chronic cough and sputum production; persistent infection with typical CF pathogens including Staphylococcus aureus, Haemophilus influenzae, Pseudomonas aeruginosa, and Burkholderia cepacia;
radiographic evidence of bronchiectasis; and chronic sinusitis, often with nasal polyposis.
Gastrointestinal tract and nutritional abnormalities as manifested by pancreatic insufficiency or recurrent pancreatitis, meconium ileus, or distal intestinal obstruction syndrome, failure to thrive or chronic malnutrition, and evidence of focal biliary cirrhosis.
Male urogenital problems as manifested by the congenital bilateral absence of the vas deferens as well as by obstructive azoospermia.
Individuals who demonstrate one or more of these features fulfill the criteria for a CF phenotype.
In addition to these clinical criteria, patients must have supporting laboratory evidence of CFTR protein dysfunction. This can be demonstrated in one of three ways. The first is a sweat chloride level greater than 60 mEq/L. The levels of sodium and chloride in the eccrine sweat of patients with CF are almost always elevated as much as three to six times normal. This abnormality is present from birth and persists without any relation to the severity of pancreatic or pulmonary disease. Hyponatremic collapse can occur after heat exposure and excessive sweating. The sweat test has high sensitivity and specificity and is the recommended initial screening test in those suspected of having disease.46,169 There is great heterogeneity in the clinical manifestations of CF. Some patients have all of the classic manifestations of CF from infancy, while others have much milder or even atypical disease manifestations and still carry mutations on each of the CFTR genes. Two percent of CF patients do not have elevated sweat chloride levels. CFTR genotyping demonstrating two mutations can confirm the diagnosis in those with high clinical suspicion and a negative sweat test. Over 1,400 mutations have been identified. The sensitivity of genotyping for CF depends on the number of mutations being tested for and the ethnicity of the individual.46 Last, direct in vivo measurement of CFTR function via transepithelial nasal potential difference is available at some CF centers.104 The intracellular concentration of the chloride ions results in a more negative bioelectric potential difference across CF epithelia as compared with normal tissues. The resting transepithelial elective potential is elevated from the normal range of 25 to 30 mV to 55 to 90 mV in CF individuals. Some drugs, such as amiloride, selectively inhibit the sodium pump and thus may reduce the transepithelial potential to nearly 0 mV; on the other hand, the chloride flux response, which is normally increased by antagonists such as isoproterenol, is absent in individuals with CF.105,106 The diagnosis of challenging cases may be aided by testing the nasal PD.
Nearly 90% of CF patients have pancreatic exocrine insufficiency with malabsorption of fat and fat-soluble vitamins. In many cases, despite efforts to maximize caloric intake, malnutrition results in failure to reach maximum growth and weight potential.25 The Cystic Fibrosis Foundation has set a goal body mass index of 22 for women and 23 for men.115 Studies suggest an association between nutritional status and outcomes, and recent CF management guidelines stress the importance of nutritional status.125,221
Other related conditions include delayed sexual maturation in boys, delayed menarche in girls, and rectal prolapse. Women do not have impaired fertility and have successful pregnancies as long as they have adequate pulmonary and nutritional status.133 Intussusception and meconium ileus equivalent, recurrent abdominal pain, and bowel obstruction collectively termed distal intestinal obstructive syndrome are seen in older children and adults with hard, impacted stool. Biliary tract abnormalities include an increased incidence of cholelithiasis (as high as 25%), focal biliary cirrhosis, and portal hypertension (symptomatic in 5%–10%). Severe liver disease may be unsuspected. Stern and colleagues190 reported that 8 of 61 patients who died with cor pulmonale had asymptomatic biliary cirrhosis.
There is a steady increase in the incidence of diabetes mellitus with advancing age. In one series, diabetes occurred in 32% of adults >25 years of age, and another 23% had an abnormal glucose tolerance test.118 The typical age of onset is between 18 and 24 years. Rosenecker166 and Koch108 presented evidence that insulin deficiency leads to a direct decline in pulmonary function; also that in CF patients with diabetes mellitus, the percent predicted forced expiratory volume in 1 second (FEV1) is less in all age groups as compared with those without diabetes (Table 84-1). Poorly controlled diabetes is associated with an increased risk of mortality in individuals with CF, and aggressive management of diabetes is an important component of CF care.140
Osteoporosis is regarded as a late symptomatic complication in adult CF patients, although low bone densities and fractures may be seen in children. In one series, a history of fractures during childhood was obtained in 52.8% of 70 CF patients.10 Another investigator reported an incidence of 25.1% in 143 children.82 Both of these incidences are well above the expected rate. Rib and vertebral compression fractures are the more common fractures. Bone densities (g/cm2) were found to be 35% and 26% lower in both men and women with CF, respectively, than in normal persons.13 The mechanism of this occurrence is unknown, but contributing factors to the lower bone densities seen in patients with severe pulmonary disease are malabsorption of vitamin D, calcium, and other nutrients; hypogonadism; inactivity; respiratory acidosis; and cytokines from chronic infection.146 Although severe problems are noted in only a minority of CF patients, severe osteoporosis with major kyphosis and vertebral fractures may contraindicate lung transplantation. Furthermore, any existing osteoporosis is made more severe after lung transplantation.8 Hypertrophic pulmonary osteoarthropathy and inappropriate secretion of antidiuretic hormone may occur in CF as in other types of chronic pulmonary disease.
Table 84-1 Mean Value of Percent Predicted FEV1 in Cystic Fibrosis Patients | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||
Most CF patients develop chronic sinusitis. The maxillary and ethmoid sinuses are opacified in 90% to 100% of children >8 months old; the frontal sinuses rarely develop. Most patients present with symptoms of nasal obstruction and purulent rhinorrhea between 5 and 14 years of age. Treatment is with oral antibiotics and nasal steroids. Twenty percent of CF patients require surgical treatment.154 Nasal polyposis is commonly associated, although this may resolve by late adolescence. Some have suggested that all CF patients who are candidates for lung transplantation should have operative drainage of the maxillary sinuses because they are typically contaminated with Pseudomonas aeruginosa and are a source of pulmonary infection.122 Others believe that intensive medical therapy is better than invasive procedures.226 A recent study suggests that surgical therapy after transplantation leads to decreased rates of colonization of the lower respiratory tract.86 The controversy continues. When lung transplantation is planned, some centers perform operative drainage routinely,238 whereas others do so infrequently.135
Genetics of Cystic Fibrosis
An explosion in research and knowledge about the genetics and molecular and cellular biology of CF has occurred since 1989. Rommens,164 Riordan,162 and B.-S. Kerem99 and their associates isolated and cloned the CF gene; identified and characterized CFTR, the protein product of the gene; and pinpointed the DNA sequence of the defective gene involved with most cases. The normal gene is composed of 250,000 nucleotide base pairs and is located on the long arm of chromosome 7 (7q31.3). The gene is transcribed into a 6.5-kb messenger RNA that encodes a 1480–amino acid protein. Tsui205 reviewed the nearly 300 different gene mutations that had been reported to the Cystic Fibrosis Genetic Analysis Consortium Database, but this was only the tip of the iceberg. Over 1,400 mutations have been discovered so far.41 The genotype–phenotype relationship is complex and is affected by pollution, smoking, bacterial infection, and malnutrition as well as by various therapeutic agents.
Although >1,000 CF-causing mutations have been described, only 22 have been identified with a frequency of at least 0.1% of known alleles. The remaining mutations are very rare and limited to a few individuals. The most common mutation is a 3-bp deletion that codes for phenylalanine at position 508 of the CFTR protein, delta F508. Delta F508 is found in 67% of white patients and is the most severe genetic lesion for homozygotes.114 An affected individual will have two abnormal CF genes and may be homozygous or heterozygous for the delta F508 allele or a nondelta F508 allele. Carriers of only one gene are usually asymptomatic; however, increased airway reactivity and an increased incidence of wheezing in heterozygous parents of children with CF has been documented.45 A group of 25 infertile men with bilateral absence of the vas deferens, none of whom had pulmonary symptoms, were investigated: 13 were carriers for delta F508 and 3 had a delta F508/nondelta F508 genotype.5 The delta F508 allele and those alleles whose DNA sequencing is known can be detected by specific DNA testing, allowing accurate detection of carriers and of patients who have atypical or minimally symptomatic disease. Such identification is difficult if the alleles can be identified only by their relationship to known DNA markers and with comparison to the DNA of affected family members. The delta F508 allele is the most common CF gene found in white patients of northern European descent, especially Danish patients, and is found less frequently in southern European, black, and Ashkenazi Jewish patients. The other 21 common mutations are found in higher frequency in these ethnic groups.
Several studies have shown that different alleles appear to influence the clinical severity of disease, particularly of the pancreas.101,113 As noted, patients who are homozygous for delta F508 tend to have severe classic CF and almost all have pancreatic insufficiency. As many as 10% of the nondelta F508 alleles, however, lead to a mild form of CF in which the patient has pancreatic function sufficient for normal digestion. These al- leles appear to be dominant over delta F508. There is no good correlation of genotype with the degree of lung disease or liver disease, and no study was able to correlate pulmonary function testing with genotype unless the patients were subdivided according to pancreatic function.99,172 Individuals with pancreatic sufficiency tend to have better pulmonary function, perhaps on the basis of better nutrition.38,69
Mutations in the CF gene can disrupt CFTR function in epithelial cells in many ways, from complete loss of protein to poor chloride conductance. Zeitlin225 classified the basic defect of CFTR into five categories:
Class I mutations are premature transcription terminations causing a truncated nonfunctional CFTR, resulting in complete absence of protein expression.
Class II missense mutations consist of aberrantly folded CFTR protein that is degraded by the cell quality-control system.
Class III mutations lead to defective regulation of the CFTR protein and, consequently, the absence of CFTR function.
Class IV mutations lead to chloride channel conduction defects.
Class V mutations result in reduced amount of functional CFTR protein due to abnormal gene splicing.
Examples of gene mutations, resulting defects, and possible general therapeutic modalities for these classes are listed in Table 84-2.
The development of a cure for CF must involve the alteration of the mutant CFTR or the transfer of one normal CF gene into enough respiratory epithelial cells to overcome the basic cellular defect. The CFTR protein has been purified and reconstituted in an in vitro system.17 New forms of treatment can be evaluated in mouse models in which the CFTR protein has been inactivated.55,183 Because CF is a recessive disorder determined by a lack of gene function, adding one normal gene to the nuclei of the appropriate cells should convert the cell to a carrier state and allow normal function. Only 5% of normal gene expression should be necessary.
The normal CF gene has been delivered directly into respiratory epithelial cells using adenoviruses and retroviruses as vectors. First, DNA material that allows the virus to replicate within the infected cell is removed; then, the foreign gene for CFTR production is inserted into the viral genome. Large quantities of the replication-deficient recombinant virus can be produced in cell culture. Rosenfeld and coworkers167 showed that CFTR protein is expressed within the airway epithelial cells of rats for ≤6 weeks after infection with an adenovirus vector. The adenovirus can transfer the gene into differentiated tissues that are not undergoing frequent cell division, but the effect on the respiratory cells may last only several weeks. Recombinant
retroviruses have been used to transform cells in vitro to express normal CFTR channel function.56 Human studies using adenoviral and nonviral cationic lipid delivery systems demonstrate gene expression in upper and lower airway epithelium.2,31,106 However, gene-transfer therapy has unfortunately been plagued with poor efficiency, short duration of gene expression, and immune and inflammatory responses to the adenoviral vectors.220,223 Although these initial and subsequent studies are encouraging, improvement of gene delivery is required prior to widespread use of this treatment. There are also ongoing investigations into development of therapeutic strategies to correct or partially correct the abnormal CFTR protein function based upon the class of gene mutation. Although some phase I/II human trials are promising, to date there are no phase III trials documenting clinical efficacy.
retroviruses have been used to transform cells in vitro to express normal CFTR channel function.56 Human studies using adenoviral and nonviral cationic lipid delivery systems demonstrate gene expression in upper and lower airway epithelium.2,31,106 However, gene-transfer therapy has unfortunately been plagued with poor efficiency, short duration of gene expression, and immune and inflammatory responses to the adenoviral vectors.220,223 Although these initial and subsequent studies are encouraging, improvement of gene delivery is required prior to widespread use of this treatment. There are also ongoing investigations into development of therapeutic strategies to correct or partially correct the abnormal CFTR protein function based upon the class of gene mutation. Although some phase I/II human trials are promising, to date there are no phase III trials documenting clinical efficacy.
Table 84-2 Organization of Regular Mutations in Transmembrane Conductance in Cystic Fibrosis According to Defect and Therapeutic Modality | |||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||
Pathophysiology of Pulmonary Disease
The gross and histopathologic findings of the CF lung reflect the effects of the cellular defect and chronic infection. The earliest histologic changes appear in the tracheobronchial submucosal glands, which become obstructed and dilated. These glands are the main site within the airway for the expression of the CF cell protein. Thereafter, thick mucus obstructs bronchioles and bronchi in a scattered fashion throughout the lungs. Bacterial colonization and mucosal infection follow.61 Infection is followed by an intense neutrophilic response. There is persistent inflammation with elevated interleukin-8 (IL-8) and neutrophil elastase. As neutrophils break down, they release large amounts of high-molecular-weight DNA, which increases the viscosity of endobronchial secretions and impairs mucociliary clearance.131 Ciliated cells are destroyed as bronchial epithelium undergoes metaplasia; the submucosa is infiltrated by inflammatory cells, with submucosal abscess formation; and adjacent lymphoid tissue proliferates. The proteolytic destruction of bronchial tissue results in fibrosis, with weakening of the integrity of bronchial walls and progressive bronchiectasis. The incidence of bronchiectasis rises steadily after 1 year of age.19 Airway volume, as opposed to parenchymal volume, is increased with progressive disease. The airway volume in the CF patient is 10% to 20% of the total lung volume, as opposed to 4% in the normal individual.300 This increased proportion of bronchial volume and the occurrence of cystic lesions is seen primarily in the upper lobes. Some have speculated that this difference may reflect a decrease in the clearance of secretions and their proteolytic contents from the upper lobes, perhaps because of a smaller excursion of the upper lobes during ventilation.224 In CF patients the inflammation is centered in the airways. There is a decrease in the density of small airways (airways <2 mm/cm2) that occurs with increasing age, and the percentage of the smallest airways (airways <0.35 mm) is lower in patients with hypercapnia.80
Individuals with CF often demonstrate characteristic bacterial flora in their sputum, with chronic infection with Staphylococcus aureus and Haemophilus influenzae as children and Pseudomonas aeruginosa as adults. Approximately 80% of individuals with CF are chronically infected with P. aeruginosa by the age of 18 years.40 There is a variable but persistent decline in pulmonary function that occurs after P. aeruginosa colonization of the respiratory tract.103 P. aeruginosa adheres to damaged or abnormal mucosa through specific interactions between bacterial adhesions and the cell surfaces. The mucosa may be damaged by early childhood infection with viruses, S. aureus, and H. influenzae.21,150,196 Viral infections are important in the exacerbation of bacterial infection and the decline in pulmonary function.153 The abnormally viscid CF secretions impede mucociliary clearance of bacteria. P. aeruginosa isolates from the lungs of CF patients are distinct from those causing acute infections in other settings. Early isolates have phenotypes typical of environmental strains. Over time, a number of mutations occur in P. aeruginosa in the CF lung environment that promote persistence of the organism. These include loss of flagella and pilli, loss of O-side chains on LPS, and gain of an alginate coat that protects against phagocytosis in mucoid strains. These altered isolates are more likely to have multidrug antibiotic resistance. In CF, P. aeruginosa isolates grow in biofilms—sessile bacterial communities that form in aggregates on surfaces through synthesis of a hydrated polymeric matrix. Within biofilms, noninvasive colonies of mucoid organisms are slow-growing and protected from the action of antibiotics, immunoglobulins, and neutrophils.70 Complex interactions occur between the bacteria and the large numbers of antibodies, immunoglobulins, and neutrophils that are attracted to the site of infection. Both neutrophils and P. aeruginosa organisms release elastase and other
proteolytic enzymes that are destructive to bronchial connective tissue. The deleterious effects of neutrophil elastase include stimulation of mucosecretory differentiation of respiratory epithelium, with increased secretion of mucus; destruction of structural fibers of the airway; inhibition of phagocytosis; and inactivation of antipseudomonal antibodies. The predisposition to Pseudomonas infection in patients with CF may be due in part to impaired clearance directly related to the defect in CFTR.152 CFTR knockout mice have an impaired ability to control Pseudomonas lung infections.74 There is increased binding of P. aeruginosa to respiratory epithelial cells, which can be reversed by CFTR gene transfer.44 The rates of multidrug-resistant (resistance to more than three antimicrobial agents) P. aeruginosa has increased from 7.2 in 2001 to 9.9% in 2003.96
proteolytic enzymes that are destructive to bronchial connective tissue. The deleterious effects of neutrophil elastase include stimulation of mucosecretory differentiation of respiratory epithelium, with increased secretion of mucus; destruction of structural fibers of the airway; inhibition of phagocytosis; and inactivation of antipseudomonal antibodies. The predisposition to Pseudomonas infection in patients with CF may be due in part to impaired clearance directly related to the defect in CFTR.152 CFTR knockout mice have an impaired ability to control Pseudomonas lung infections.74 There is increased binding of P. aeruginosa to respiratory epithelial cells, which can be reversed by CFTR gene transfer.44 The rates of multidrug-resistant (resistance to more than three antimicrobial agents) P. aeruginosa has increased from 7.2 in 2001 to 9.9% in 2003.96
The concept of bronchial tissue destruction is supported by laboratory and pathologic evidence.29 Uninhibited elastase activity is indicated by the increased urinary excretion of amino acid degradation products of elastin and by the presence of fragmented elastin fibers in the CF lung at autopsy. Chronic mucosal infection, excessive production of viscid secretions, and the progressive destruction of bronchial tissue are self-perpetuated in a vicious cycle as the inflammatory and hyperimmune response from the host continues unabated. Chemical evidence of this response is the hypergammaglobulinemia, increased numbers of immune complexes in sputum and serum, and the increase in circulating P. aeruginosa antibodies found in patients with progressively severe lung disease and clinical deterioration.
Later in the course of disease, other organisms are often identified. These include B. cepacia, Stenotrophomonas maltophilia, Achromobacter xylosoxidans, Aspergillus, and nontuberculous mycobacteria. Of these organisms, the most serious is B. cepacia complex. B. cepacia syndrome is heralded by high fever, bacteremia, and necrotizing pneumonia; it is generally fatal. Lewin reviewed the course of patients colonized with this organism at several CF centers. Earlier clinical deterioration and death occurred in those who also had moderate or advanced lung disease.122 B. cepacia is now recognized as a group of nine closely related species termed genomovars. The majority of CF infections with B. cepacia complex are caused by genomovars II (Burkholderia mulivorans), III (Burkholderia cenocepacia), and V (Burkholderia vietnamiensis).34,126 In addition to being virulent, B. cepacia complex is often multidrug-resistant. The presence of these organisms may affect outcomes after lung transplantation and are considered a relative contraindication at most U.S. transplant centers.
Pulmonary Function Studies
Spirometry and plethysmography are the principal measures of pulmonary status in individuals with CF >5 years of age. Serial measurements are used to document stability or progression of airway obstruction, acute changes associated with pulmonary exacerbations, and response to therapy. Most children can cooperate with testing by age 5 to 6, and pulmonary function studies are performed at regular intervals thereafter. The early and scattered obstruction of peripheral airways causes predictable abnormalities in arterial oxygenation, pulmonary volumes and capacities, and airflow mechanics. Early disease is confined to the small airways, and maximum expiratory airflows are decreased initially only at small lung volumes. Therefore, the maximum expiratory airflow after expiration of 75% of vital capacity (Vmax75%) and the average maximum expiratory flow during the middle 50% of vital capacity are more sensitive than the FEV1.212 Another early finding may be elevated residual volume (RV) and an increased ratio of RV to total lung capacity (RV/total lung capacity) consistent with gas trapping. As patients develop progressive obstructive lung disease, FEV1 begins to decline. The progressive loss of lung function begins in the teens and averages between 1% and 4% per year. Also, bronchial hyperresponsiveness occurs in about half of patients with CF.138 Different rates of decline in lung function results in a wide spectrum of severity of lung disease in adults, with approximately 25% having severe lung disease, 40% with moderate disease, and 35% with mild disease or normal lung function.40 FEV1 is the most widely used parameter of pulmonary status in CF for several reasons. Multiple epidemiologic studies have demonstrated that FEV1 is a marker of disease progression and a predictor of mortality.102,131 In a large observational study, predictors of the rate of decline in FEV1 include poor nutritional status, infection with P. aeruginosa, persistent crackles on examination, and frequent pulmonary exacerbations.111 Additionally, FEV1 is the primary outcome measure used to define clinical efficacy for new therapeutic modalities.155 In advanced disease, forced vital capacity (FVC) and total lung capacity (TLC) decline because of progressive scarring, air trapping, and increased dead-space ventilation. As bronchiectasis and airway obstruction become more pronounced, ventilation/perfusion mismatch leads to hypoxemia. Hypercapnia occurs relatively late in the course of CF lung disease and is often a preterminal finding. Chronic hypoxemia and hypercarbia may lead to muscular hypertrophy of the pulmonary vasculature, right ventricular hypertrophy, and eventually cor pulmonale with right heart failure.
Radiographic Findings
The earliest radiographic abnormalities in CF are evidence of hyperinflation with flattening of the domes of the diaphragm and an increased anteroposterior diameter of the chest. This may occur as early as infancy. Bronchovascular markings are prominent, especially in the upper lobes. Atelectasis of the right upper lobe, right middle lobe, or left lower lobe in infants and young children should suggest the diagnosis of CF. The chest radiograph of a patient with well-established disease reveals a diffuse cystic interstitial process with maximum involvement of the upper lung fields (Fig. 84-1). The most frequently occurring abnormalities include hyperinflation, usually of the upper lobes; patchy linear and nodular densities, probably representing bronchiectasis or small peribronchial abscesses; and lobar segmental atelectasis (Fig. 84-2). Apical blebs appear in older patients.27 Chest radiographs are useful in defining disease progression and less sensitive in demonstrating changes during acute exacerbation. Chest radiograph scoring can predict pulmonary function in children. In adults, there is a correlation between chest radiographs and pulmonary function, especially the FEV1. Changes in the radiograph, however, lag behind decreases in FEV1.165
High-resolution CT (HRCT) of the chest is more sensitive than chest radiography in detecting changes such as air
trapping and airway wall thickening in early disease.22 HRCT can be used to evaluate for localized disease and clarify the extent of bronchiectasis.47 HRCT changes may precede changes in pulmonary function.48 HRCT is often used clinically to follow disease progression and evaluate for response to therapy following acute pulmonary exacerbation. (Shaw97). However, there are no consensus guidelines for routine use of HRCT in CF care.
trapping and airway wall thickening in early disease.22 HRCT can be used to evaluate for localized disease and clarify the extent of bronchiectasis.47 HRCT changes may precede changes in pulmonary function.48 HRCT is often used clinically to follow disease progression and evaluate for response to therapy following acute pulmonary exacerbation. (Shaw97). However, there are no consensus guidelines for routine use of HRCT in CF care.
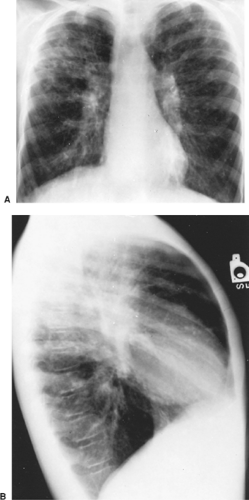 Figure 84-1. Anteroposterior (A) and lateral (B) radiographs of a 12-year-old boy show hyperexpansion of the chest wall and depressed diaphragms from overinflated lungs. Linear and patchy densities are concentrated in a contracted right upper lobe. Note the normal heart size and the prominent pulmonary arteries. The vertebral column is osteoporotic. |
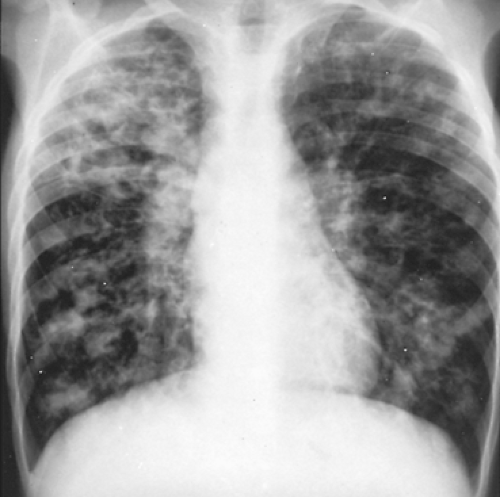 Figure 84-2. Nodular cystic changes (bronchiectasis and scarring) and linear densities are prominent in the radiographs of a 16-year-old with more severe disease. |
Comprehensive Medical Treatment
There are several comprehensive consensus statements regarding optimal care of CF patients.66,221 The Cystic Fibrosis Foundation has established guidelines that must be met if a facility is to become a certified adult CF care center, including the requirement that the center have one or more physicians with expertise in adult CF care and a dedicated multidisciplinary team. Outcomes for CF patients are improved if they are cared for at such centers.128 Maintaining pulmonary health by optimizing airway secretion clearance, treatment of obstructive lung disease, anti-inflammatory therapy, and antibiotics are among the cornerstones of CF care.
Methods to optimize clearance of airway secretions include chest physiotherapy, mucolytics, and hyptertonic saline. Segmental postural drainage, chest percussion or vibration, breathing exercises, and active aerobic exercise all contribute to the mechanical removal of secretions.84 There are no randomized controlled trials in stable patients with CF comparing chest physiotherapy with spontaneous cough; however, the long-term benefits of daily maintenance chest physiotherapy are believed to be helpful in improving airway clearance.205 One controlled study reported that standard chest physiotherapy improved sputum production and lung function in the setting of acute pulmonary exacerbation.26 Several active sputum clearance–assist devices are available, including oral airway oscillators such as Acapella devices. There are also passive clearance-assist devices, such as high-frequency chest wall oscillators. Small studies have demonstrated mild improvement in FEV1 or clinical scores with some of these devices.73,87 With limited outcomes data, the choice of technique depends on patient satisfaction and compliance.
Mucolytics promote sputum clearance by decreasing sputum viscosity. DNA, released by degenerating inflammatory cells,
further increases the viscosity of mucus. Purified recombinant human deoxyribonuclease, DNAse I (Pulmozyme), can degrade the large DNA molecules into smaller strands.180 Thinning of the sputum and improvement in pulmonary function testing was demonstrated in an early study in 16 CF patients after 1 week of aerosol treatment.88 Several recent randomized controlled trials have demonstrated significant improvement in FEV1 and reduction in pulmonary exacerbations with daily use of nebulized Pulmozyme. In one trial, where Pulmozyme 2.5 mg nebulized daily was compared with placebo, FEV1 increased by 5.8%, and exacerbations requiring intravenous antibiotics decreased by 22%.67 Chronic daily therapy with nebulized Pulmozyme is recommended for all adult patients with CF with mild to severe pulmonary dysfunction.66 Aerosol and oral treatments with the mucolytic agent N-acetyl-L-cysteine have shown no clinical benefit or improvement in lung function in CF and are not currently recommended.66 Hypertonic saline increases hydration of airway surface liquid, thus improving the clearance of secretions.54 Several controlled trials of nebulized 6% to 7% saline twice daily have had variable results. In one study of 58 patients, treatment with hypertonic saline led to a 15% increase in mean FEV1.60 Another larger study revealed no difference in FEV1 compared with placebo but did demonstrate an impressive 56% reduction in pulmonary exacerbations requiring antibiotics.59 The most common reported side effect is acute, transient bronchospasm and patients are routinely pretreated with a beta agonist. Nebulized hypertonic saline twice daily is recommended for all adult CF patients with mild to severe pulmonary dysfunction.66,210
further increases the viscosity of mucus. Purified recombinant human deoxyribonuclease, DNAse I (Pulmozyme), can degrade the large DNA molecules into smaller strands.180 Thinning of the sputum and improvement in pulmonary function testing was demonstrated in an early study in 16 CF patients after 1 week of aerosol treatment.88 Several recent randomized controlled trials have demonstrated significant improvement in FEV1 and reduction in pulmonary exacerbations with daily use of nebulized Pulmozyme. In one trial, where Pulmozyme 2.5 mg nebulized daily was compared with placebo, FEV1 increased by 5.8%, and exacerbations requiring intravenous antibiotics decreased by 22%.67 Chronic daily therapy with nebulized Pulmozyme is recommended for all adult patients with CF with mild to severe pulmonary dysfunction.66 Aerosol and oral treatments with the mucolytic agent N-acetyl-L-cysteine have shown no clinical benefit or improvement in lung function in CF and are not currently recommended.66 Hypertonic saline increases hydration of airway surface liquid, thus improving the clearance of secretions.54 Several controlled trials of nebulized 6% to 7% saline twice daily have had variable results. In one study of 58 patients, treatment with hypertonic saline led to a 15% increase in mean FEV1.60 Another larger study revealed no difference in FEV1 compared with placebo but did demonstrate an impressive 56% reduction in pulmonary exacerbations requiring antibiotics.59 The most common reported side effect is acute, transient bronchospasm and patients are routinely pretreated with a beta agonist. Nebulized hypertonic saline twice daily is recommended for all adult CF patients with mild to severe pulmonary dysfunction.66,210
Treatment of obstructive lung disease is important in CF patients. Several trials have evaluated the role of bronchodilators in CF. Most studied have focused on inhaled beta2 agonists and anticholinergics. A Cochrane review in 2005 evaluated 14 of these publications. The authors concluded that both short- and long-acting beta2 agonists can be beneficial both in the short and long term in individuals with demonstrable bronchodilator responsiveness. They found insufficient evidence, however, to support the routine use of inhaled anticholinergic bronchodilators such as ipratropium bromide.66,78 Beta2 agonists are also recommended for use immediately prior to sessions of chest physiotherapy, in order to facilitate clearance of airway secretions, and prior to inhaled hypertonic saline and inhaled antibiotic treatments to prevent bronchospasm.
As detailed previously, there is an excessive inflammatory response in the airways of CF patients. Several clinical trials of anti-inflammatory therapy have demonstrated benefit; however, adverse effects have limited the use of many of these agents. Medications investigated include ibuprofen, oral corticosteroids, and inhaled corticosteroids. Ibuprofen was shown to ameliorate the inflammatory response to lung infection in a rat model.109 In one study, high-dose ibuprofen (20–30 mg/kg up to 1,600 mg twice a day) slowed the progression of pulmonary disease in mildly affected patients (FEV1 >60% predicted), particularly in children aged 5 to 12 years.110 In adults, the occurrence of gastrointestinal bleeding, increased risk of hemoptysis, and renal toxicity may outweigh any benefit of the drug. A small short-term trial of piroxicam, a nonsteroidal anti-inflammatory drug (NSAID), versus placebo failed to demonstrate any improvement in lung function.185 Twice daily use of ibuprofen is recommended in CF patients >6 years of age with FEV1 over 60% predicted.66,117 There have been three large randomized controlled trials evaluating the use of empiric oral cortico- steroid use in children. All used alternate-day steroids at doses of 1 to 2 mg/kg. The duration of use varied from 3 weeks to 4 years. Two of the studies reported significant improvement in pulmonary function with corticosteroid use.12,75 A third study found an unacceptable incidence of complications—such as cataracts, growth failure, and glucose intolerance—and no significant change in pulmonary function.58 In adults, a randomized controlled trial using prednisolone 30 mg daily for 3 weeks demonstrated no change in lung function; no adverse events were reported. Although corticosteroids do appear to improve lung function in children, their routine use is not recommended owing to severe adverse effects.66,70 There are insufficient data for the routine use of corticosteroids in adults or for their use as adjunctive therapy in acute pulmonary exacerbations of CF.66 The efficacy of inhaled corticosteroids has been evaluated in several randomized controlled trials in both children and adults. No study demonstrated statistically significant improvement in lung function (FEV1) or decreased risk of pulmonary exacerbation; therefore they are not recommended for routine use.14,66 Although both systemic and inhaled corticosteroids are not recommended for routine use, their use may be appropriate in CF patients with severe asthma and/or allergic bronchopulmonary aspergillosis. Other anti-inflammatory agents currently in early trials include glutathione, S-nitrosoglutathine, and leukotriene modifiers.
Antibiotics have several roles in the treatment of CF. They may have a role early to eradicate primary P. aeruginosa infection. They are used chronically to slow the decline in pulmonary function and decrease frequency of pulmonary exacerbations and acutely in pulmonary exacerbations to alleviate symptoms and restore pulmonary function. Macrolides, inhaled antipseudomonal antibiotics, and intensive intravenous regimens for acute pulmonary exacerbations are the types of antibiotics prescribed most frequently in CF.
There is a growing body of evidence supporting the chronic use of macrolide antibiotics in CF. The mechanism of benefit may be related to anti-inflammatory or antimicrobial properties. Macrolide antibiotics have been used with good effect in diffuse panbronchiolitis, a disease process with some features similar to those of CF.112 This experience has led to interest in the use of macrolides in CF. The largest randomized clinical trial studied 185 patients who were chronically infected with P. aeruginosa and had an FEV1 >30%.171 Patients received azithromycin 500 mg PO or placebo 3 days a week for 24 weeks. Patients taking azithromycin had a 4.4% improvement in FEV1, while those receiving placebo had a 1.8% reduction. There were 40% fewer pulmonary exacerbations in the treatment group. Benefit has been demonstrated in multiple studies; therefore thrice weekly azithromycin is recommended for all CF patients >6 years of age with P. aeruginosa–positive sputum cultures.66,186 The benefit of macrolides in children <6 years, in patients with B. cepacia, and in those not infected with Pseudomonas is currently undefined; studies are ongoing. Prior to initiating macrolide therapy, a sputum culture should be evaluated for nontuberculous mycobacteria; in their presence, the use of macrolides may result in the induction of macrolide resistance.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


