Pulmonary Arteriovenous Malformations
HISTORY
Pulmonary arteriovenous malformations (PAVMs) were first described in the late 19th century; Churton1 reported the autopsy findings in a young boy with cyanosis in 1897. Based on the correlation of physical with postmortem findings, the triad of cyanosis, clubbing, and polycythemia was identified with PAVM in 1932.2 Hereditary hemorrhagic telangiectasia (HHT) was first connected to PAVM in 1938.3 As described below (Causes and Disease Associations), HHT is often intimately related to PAVMs—a fact that prompts the following discussion of the history of HHT.
Hereditary epistaxis was first described in 1864,4 though neither that nor Babbington’s description a year later report an association with telangiectasia.5 These reports were not generally recognized; nor were subsequent descriptions of telangiectasia, hereditary transmission, and epistaxis by Legg6 in 1876, or a similar kindred reported by Chiari in 1887.7 The first widely recognized connection of epistaxis to telangiectasia was made by Rendu in 1896.8 Osler9 added three cases, and recognized familial occurrence in 1901. Weber10 elucidated the familial nature and lack of coagulation abnormality, and thus earned his eponymic association. By precedence of description, this eponym should be Rendu–Osler–Weber, even though Osler–Weber–Rendu is the most common usage. Hanes11 was responsible for naming the syndrome HHT, the designation now most often preferred, in 1909.
GENETICS
The genetic basis, if any, of isolated PAVMs remains unknown. HHT is an autosomal dominant disease. Its frequency was believed until relatively recently to be less than 3 per 100,000 people.12 Newer studies suggest a much higher prevalence. The highest frequency reported, 1:1331, occurs in the Afro-Caribbean population of the Netherlands Antilles, presumably due to a founder effect.13 Other estimates vary geographically; 1:6410 in Denmark,14 1:8000 in Japan,15 and 1:16,500 in Vermont.16 Phenotypic variation is extreme, ranging from asymptomatic to severely symptomatic, and from cases with no or few mucocutaneous lesions to those with diffuse cutaneous telangiectasias. For many patients, the disease remains undiagnosed by their primary care physicians, suggesting that disease frequency may be greater than reported, and that some patients with “isolated” PAVMs may actually have HHT.
A gene for HHT was first localized to chromosome 9, region q33–34 (9q33–34).17–19 Investigation revealed the protein product to be endoglin, which associates with the transforming growth factor-beta (TGF-β) bone morphogenetic protein (BMP) receptor complex and binds TGF-β-1 and -3.20 The same work showed the disease to be genetically heterogeneous, with multiple mutations in the responsible gene. It rapidly became clear that there were other chromosomal mutations resulting in the same syndrome, and the endoglin mutation disease was designated HHT-I; it was noted to be associated more often with PAVMs than were those with non-9q3 mutations.21,22 A haploinsufficient mouse model also demonstrated phenotypic heterogeneity which was very dependent on the genetic background.23
The activin receptor–like kinase 1 gene (ALK-1 or ACVRL1) on chromosome 12 is the second locus for HHT.24,25 It produces a TGF-β superfamily type I receptor. Mice heterozygous for a loss-of-function mutation in ALK-1 develop age-dependent vascular lesions in the skin, extremities, oral cavity and in the lung, liver, intestine, spleen, and brain, similar to those seen in HHT patients.26 Disease resulting from mutations in this gene has been designated HHT-2.
A small number of patients with juvenile polyposis also have HHT. This is due to mutations in MADH4 (encoding SMAD4); SMAD proteins influence the cellular response to TGF-β through interactions with other SMADs as transcription factors.27,28
A fourth gene abnormality producing clinical HHT in one family has been described on chromosome 5. The gene product is as yet unidentified.29
A fifth genetic abnormality in a family with HHT has been described on the short arm of chromosome 7.30 The gene product of this mutation is also unknown at present.
Most HHT appears to be caused by mutations in endoglin and ALK-1. Mutations can be identified in up to 88% of affected individuals31,32; in one series, 61% were in endoglin, 37% in ALK-1, and 2% in MADH4.33 ALK-1 mutations appear to be more common in France and Italy, with endoglin mutations more frequent in northern Europe and North America.31,32,34 PAVMs are more frequent and on the average of larger size in HHT-1.35
Genetic testing for mutations in endoglin, ALK-1, and MADH-4 is currently available from six laboratories in North America, and a number of other laboratories in Europe. An up-to-date list with contact information is maintained at http://hht.org. The primary role for testing is to identify a mutation in an index case who meets criteria for a diagnosis of HHT. If this is possible, the index case’s children and other first-degree relatives may be screened. Those with negative tests may be reassured, and those with positive tests may be evaluated for complications of HHT. On occasion, a diagnosis may be confirmed in an index case when clinical criteria are insufficient for clinical diagnosis. As with all genetic diseases, testing should be accompanied by genetic counseling.
PATHOPHYSIOLOGY
Important pathophysiologic considerations of PAVM are discussed below.
 STRUCTURE
STRUCTURE
The three essential structural elements of PAVMs are the arterial supply (“feeder vessel”), a draining vein, and the intervening aneurysmal sac. Because of this simple relationship, the label of “AVM” is a bit of a misnomer as these malformations are more characteristic of a fistulous connection between arterial and venous branches without a customary intervening capillary network that is vital for gas exchange. PAVMs appear to develop between precapillary arterioles and venules, with intervening epithelial dysplasia.36,37
Approximately 80% of PAVMs have a single feeding and a single draining vessel; the remaining 20% are complex, with two or more of each.38 By far the most common form of PAVM has a pulmonary arterial supply and pulmonary venous drainage.39 In one series, 60 of 63 PAVMs had a pulmonary arterial blood supply40 but arteries from the systemic circulation can also be involved, including arterial branches from the internal mammary artery, intercostal arteries, and subdiaphragmatic arteries. While systemic “feeders” are prone to develop as sequelae of chronic suppuration in the lung (e.g., sequestration), postpulmonary infarction after pulmonary embolism, or postembolization of pulmonary artery-to-vein malformations,41 this chapter will focus on the classic form PAVMs involving the pulmonary arteries.
 PATHOGENESIS
PATHOGENESIS
Our evolving understanding of PAVM development was summarized in a recent and thorough review of HHT.42 Briefly, pathogenesis of PAVMs is presumed to initiate from periods of increased angiogenic activity, possibly triggered at sites of vascular injury,43 and likely spurred on by an imbalance between proangiogenic signaling and reduced antiangiogenic activity. Angiogenesis is dysregulated due to altered expression of TGF-β–mediated pathways in endothelial cells of the pulmonary circulation, which plays a critical role in endothelial cell homeostasis. Mutations in one of the HHT-causative genes (described earlier) leads to either altered ligand-receptor interaction at the endothelial cell surface (ENG or ACVR1) or intracellular signaling (SMAD4) within endothelial cells. Downregulation of TGF-β expression is postulated to permit excessive endothelial cell proliferation and increase blood vessel formation44 (under the influence of proangiogenic signals from molecules such as vascular endothelial growth factor [VEGF]), form persistent direct arteriovenous connections,45 and destabilize vessels due to interactions between endothelial and mural cells (e.g., pericytes, smooth muscle cells).46 The lack of a capillary network within a PAVM and the direct communication between arterioles and venules exposes thin-walled conduits to arterial blood flow and increased shear forces. Again, genetic defects and dysregulated angiogenesis lead to a muted compensatory response within these “arterialized” veins that must dilate. Furthering this concept, work in an animal model of HHT suggests a two-step dysregulatory sequence culminating in AVM formation, whereby the initial endothelial cell proliferation is mediated by HHT-causing mutations44 and the subsequent dilation and persistence of downstream arteriovenous communications is independent of the mutations and, indeed, may be a homeostatic response to altered upstream blood flow pattern.47 Over time, these initial microscopic communications grow into macroscopic communications48 notable for relatively increased flow and passage of material normally sequestered or cleared in the pulmonary microcirculation (e.g., air bubbles, thrombi, bacteria). Further work in this area should clarify the molecular steps from altered gene expression to dysregulated multistep angiogenesis, discover precipitants of the dysregulated angiogenesis, and identify comediators, associated pathways, and environmental factors that influence angiogenesis.
Growth rates of AVMs remain unknown but is likely subject to intersubject variability and overall blood flow. One observed phenomenon of PAVM growth is accelerated growth during pregnancy leading to potential peripartum hemorrhagic complications,49 which in part is theorized to occur due to increased blood flow and higher cardiac output of pregnancy but customary hormonal alterations of pregnancy could also be influential (Fig. 75-1).50
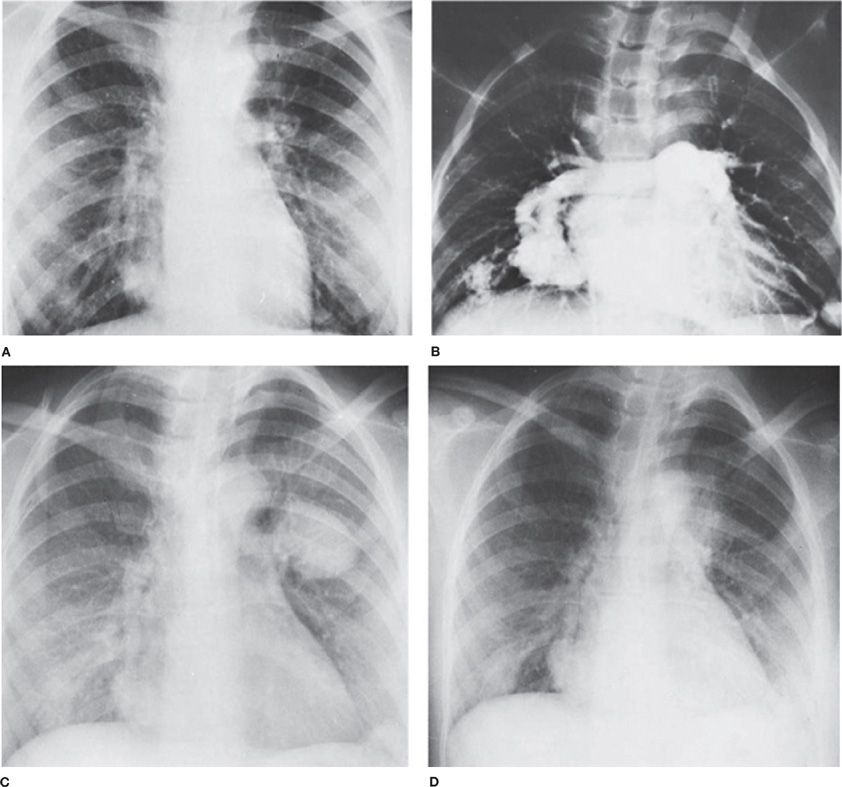
Figure 75-1 Pulmonary arteriovenous fistulas in a pregnant 24-year-old woman with hereditary hemorrhagic telangiectasia. A. Before pregnancy. Small nodular densities are seen at both bases and in the left hilus. The shunt was estimated to be 49% of the cardiac output. B. Arteriogram before pregnancy demonstrates arteriovenous fistulas of both lower lobes. C. Seven months pregnant, the patient was admitted to the hospital with hemoptysis and left hemothorax. The enlargement of the arteriovenous fistulas is striking. The pregnancy was terminated. D. Two weeks after termination of pregnancy, the nodular densities have decreased in size. (Used with permission of Dr. M. Rossman.)
 NUMBER
NUMBER
In one series, more than 60% of individuals present with more than one PAVM.51 In general, multiple PAVMs correlate with HHT. A small percentage have diffuse, multilobar PAVMs that are typically bilateral and associated with marked hypoxemia.52,53
 SIZE
SIZE
PAVMs may vary from malformations too small to be seen by radiography or angiography54,55 to those greater than 5 cm in diameter.56
 LOCATION
LOCATION
Up to 65% of PAVMs are located in the lower lobes55—a phenomenon that may be due to the increased pulmonary blood flow and pressure, and subsequent “stretch” due to hydrodynamic forces. A recent small series noted less selectivity for the lower lobes in cases of “idiopathic” PAVMs.57 The lower lobe location is the likely cause of the orthodeoxia (desaturation in an upright position) and platypnea (dyspnea in an upright position) which are sometimes seen. These symptoms may also occur with cirrhosis, which has pulmonary vascular abnormalities see below “Other Associations”. Location may also account for an increase in right-to-left shunt which occurs at total lung capacity.58
CAUSES AND DISEASE ASSOCIATIONS
Early observers thought that all PAVMs were due to HHT.38 The estimates of frequency with which PAVMs are due to HHT have varied substantially, from 36% to 95%.38,40,50,59,60 Most recent series report HHT in well over 90%.
Estimates of the percentage of patients with HHT who have associated PAVMs have varied widely. Different series have reported frequencies of 15%,50 20%,61 24%,36 33%,62 49%,63 and 57%.64 Frequency of PAVMs appears to be significantly greater in cases with ENG mutations, as opposed to ACVRL1 mutations.65 Furthermore, overall PAVM detection rates are being influenced by improving imaging techniques that provide clearer resolution of the vasculature in the peripheral areas of the lung.
OTHER ASSOCIATIONS
Cirrhosis may result in diffuse small arteriovenous connections.66 Nearly all such patients have cutaneous spider angiomas. The right-to-left shunt is probably due not to true PAVMs but, rather, to vasodilation of pleural vessels, which resemble cutaneous spiders, and increased numbers of peripheral small arteriolar branches with precapillary arteriole-to-venous connections in the peripheral respiratory lobule. As many as 44% to 60% may have positive contrast echocardiography indicative of intrapulmonary shunt;67,68 many of these patients have shunt eliminated by liver transplantation. A PAVM of significant size, known as a Rasmussen aneurysm, may also develop as a result of tuberculosis.69 Metastatic thyroid carcinoma, a highly vascular tumor, may mimic pulmonary arteriovenous fistula.70 Cavopulmonary anastomosis, used in the palliation of functionally univentricular heart disease, results in pulmonary arteriovenous connections similar to those in cirrhosis, in approximately 10% of patients. The reasons are unclear.71 Rarely, penetrating chest trauma may result in subsequent PAVM.72
PRESENTATION AND COMPLICATIONS
 PRESENTATION
PRESENTATION
The occurrence and frequency of symptoms related to PAVMs depend on how the patients are found—that is, whether they present with manifestations of disease or whether they are discovered as a result of screening. When detection occurs as a result of screening in patients with HHT, between 25% and 59% are asymptomatic.50,73–75
The age at onset is usually in the third or fourth decade.38 The mean age at detection in various series is remarkably constant at 38 to 40 years.50,51,73,74 In one series, the patients ranged in age from 5 to 76 years, with a mean of 36; 26% presented at an age less than 21 years.55 PAVMs are, however, uncommon in childhood; only 4% of affected persons are under 10.78,79
Pulmonary symptoms include dyspnea on exertion, with a frequency ranging from 27% to 71%.21,40,55,80 Platypnea and orthodeoxia also may occur. Hemoptysis ranges in frequency from 4% to 18%.21,51,55 Extrapulmonary symptoms include chest pain in 6%81 and epistaxis (largely seen in HHT), ranging from 32% to 85%.37,80,82 The mean age at onset of epistaxis in HHT is 12 years, with 54% of patients presenting by age 10. Severity of epistaxis ranges from mild to severe, with up to 45 episodes per month.76 Headache is also remarkably common in HHT patients, occurring in 43%.55 Transient ischemic attack (TIA) occurs in up to 57% of patients with PAVM, and symptomatic cerebrovascular accident in 18%.55,76
Physical signs due to the PAVM itself are relatively uncommon. As many as 25% of patients may exhibit no findings at all.40 Hypoxemia, when present, is secondary to the right-to-left shunt, and may result in cyanosis and secondary polycythemia. This tends to occur in advanced disease, and has been reported in 9% to 73% (mean 30%).40,59,76 The frequency of clubbing has been reported in an average of 32%76; it is much less common in our experience.49,56 Clubbing is nearly always associated with cyanosis. Clubbing may resolve after the PAVM is removed83 or occluded. A pulmonary bruit, which is often described, is also variable; its frequency, probably influenced by selection bias, ranges from less than 10% to 58%.40,49,76,80
Telangiectasia have been reported in up to 66% of patients with PAVM, depending on the frequency of HHT.84 These small red vascular blemishes occur most frequently on the face, followed in descending order by the lips, nares, tongue, ears, hands, chest, and feet. They often increase in size and number with age, and cutaneous telangiectasias are seldom identifiable until the second or third decade.37 We have been struck by the frequency with which classic tongue and lip telangiectasias have been passed off as nonspecific blemishes by primary care physicians.
Laboratory results are nonspecific. A complete blood count may show polycythemia, although in patients with HHT, this tendency may be overshadowed by iron deficiency anemia. Anemia was present in 94 of 292 (34%) in our series. This was more often due to GI bleeding when severe. GI blood loss of variable severity was present in 65 of 292 (22%).56
The severely affected person may have arterial hypoxemia at rest; those less severely affected may have orthodeoxia documented by supine and upright arterial blood gases.85 Arterial blood gases, determined on blood samples drawn while the patient is breathing room air, followed by 100% oxygen, may reveal a significant right-to-left shunt.86
 COMPLICATIONS
COMPLICATIONS
Pulmonary and neurologic complications of PAVMs are important considerations.
Pulmonary Complications
Significant hemoptysis occurs in fewer than 10% of patients; in our most recent series, it occurred in 5 of 142 (<4%). Two of five occurred during pregnancy.56 It may be massive and life-threatening. Bronchial telangiectasias may be the cause,62 but all cases in untreated patients in our experience have been due to PAVMs. An increasingly frequent problem in recent years is hemoptysis following extensive embolotherapy after a delay of months to years. This has generally been due to postembolization bronchial collateral formation.
Hemothorax has been reported in up to 9% of patients,87 but is usually less than 2%.76 Pregnancy has been associated with hemothorax on several occasions, perhaps related to PAVM enlargement.88–90 Hemothorax may also occur without any other predisposing factors, presumably caused by rupture of large subpleural PAVMs into the pleural space.
Typically, PAVMs are associated with a normal or low pulmonary vascular resistance (PVR) as the direct arteriovenous communications are low-resistance circuits. However, pulmonary hypertension is encountered, albeit uncommonly, in the setting of PAVMs and HHT.91 Pulmonary hypertension most often develops from increased pulmonary blood flow, which occurs in HHT because of massive hepatic arteriovenous malformations that rapidly return blood to the right side of the heart and lead to a high cardiac output state. The high output can be aggravated by concomitant chronic anemia from blood loss. In this situation, the PVR will be low as the entire pulmonary circuit, not just the PAVM, dilates to accommodate increased pulmonary blood flow; but the capacitance of the circuit has limits and additional blood flow will lead to mild pulmonary hypertension. In distinction, another more devastating but rarer form of pulmonary hypertension can be seen in the setting of PAVMs, whereby the PVR is quite high due to a proliferative vasculopathy of small pulmonary arterioles.92 In these rare cases, mutations occur primarily in ACVRL1 gene and represent a form of heritable pulmonary arterial hypertension (HPAH),93 which is a form of Group I pulmonary hypertension in the Dana Point classification of pulmonary hypertension.94 When PAH patients are discovered to have ACVRL1 or endoglin mutations during the genetic workup, PAVMs should be excluded as other clues for HHT can be subtle or absent in this rare population and PAH can be detected prior to a diagnosis of HHT being made.95,96 When PAVMs coexist with significant PAH, caution must be exercised with PAVM embolization (see Treatment section) as sudden obliteration of the low-resistance circuit could significantly increase the PVR and unsettle an already fragile right ventricle or lead to other hemorrhagic complications97,98; these rare cases should be managed in a multidisciplinary fashion by individuals familiar with the management of PAVMs and PAH.
Central Nervous System Complications
The pulmonary capillary vascular bed appears to be an important filter for otherwise asymptomatic small emboli, and may also have a significant role in cleansing the bloodstream during transient bacteremia. Most neurologic complications, which occur in 8% to 12% of patients with HHT, are complications of PAVMs. In one series, 60% were due to PAVM, including brain abscess, paradoxical embolus, and hypoxemia.37,99
TIAs occur in approximately 37% of patients with PAVMs.55 PAVMs can cause symptomatic cerebrovascular accidents (Fig. 75-2); the frequency of this complication ranges from 6% to 27%.40,55,76 In our clinic, 28 of 132 patients screened by magnetic resonance imaging (MRI) had evidence of prior paradoxical embolic stroke.56 Unfortunately, paradoxical embolization to the brain may be the first manifestation of an occult pulmonary venous malformation. This has been a particularly regrettable repetitive problem in young women who smoke and take oral contraceptives. Care should be taken to avoid air embolism; IV should be free of air and a micropore inline filter used.
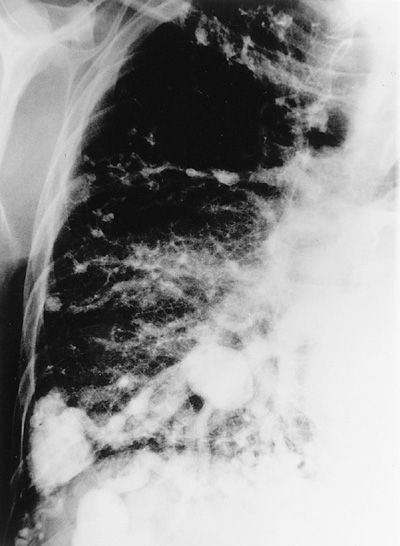
Figure 75-2 Right-sided pulmonary angiogram showing multiple PAVMs in a middle-aged man with clubbing, polycythemia, and CT evidence of several prior strokes.
Brain abscess occurs in 3% to 10% of patients with PAVMs.37,76 In a series in our clinic, 5/132 (4%) had prior brain abscess.56 Up to 1% of HHT patients may have brain abscesses (1000 times the incidence in the general population). In one series, 5 of 31 patients had recurrent abscess100; in another, 6 of 128.101 Up to 8% of brain abscesses in the general population may be due to PAVMs.102 Unfortunately, brain abscess may also be the first symptom of an occult PAVM (Fig. 75-3), and many years may elapse before diagnosis of PAVM (Fig. 75-4)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


