Pulmonary Alveolar Proteinosis Syndrome
Pulmonary alveolar proteinosis (PAP) syndrome is characterized by the accumulation of surfactant in alveoli and terminal airways resulting in hypoxemic respiratory failure.1 This fascinating syndrome continues to serve as a paradigm for disease discovery and development due to a globalized collaborative network, employment of diverse clinical, basic, and translational research approaches, and active patient involvement. While PAP occurs in many clinical settings including recently identified genetic etiologies, its molecular basis is now known in more than 90% of cases, and the molecular basis of the role of granulocyte macrophage colony–stimulating factor (GM-CSF) in surfactant homeostasis has been defined. Diseases associated with PAP can be grouped into primary PAP, secondary PAP, and congenital PAP based primarily on pathogenesis involved. Primary PAP is caused by impairment of GM-CSF–dependent surfactant clearance by alveolar macrophages and accounts for approximately 90% of all cases.2 Secondary PAP occurs as a consequence of a comorbid condition that impair surfactant clearance by alveolar macrophages and accounts for about 5% of cases.3 Congenital PAP is a clinically distinct and pathogenically heterogeneous group of genetic disorders associated with the production of abnormal surfactant and accounts for about 5% of cases.4 Because of its increased frequency and greater research attention, primary PAP will be the focus of this chapter and data for secondary and congenital PAP will be provided where available.
PATHOGENESIS
In their initial description of PAP in 1958, Rosen et al.5 established that the material accumulating within alveoli in PAP was composed of lipids, proteins, and a small amount of carbohydrate. Research over the past two decades has shown that in more than 90% of patients pathogenesis is driven by disruption of GM-CSF signaling, which blocks terminal differentiation of alveolar macrophages thereby impairing their ability to clear surfactant.2 GM-CSF is a 23-kDa cytokine produced by respiratory epithelium and other cells6,7 initially identified by its ability to stimulate the formation of macrophage and granulocyte colonies from hematological progenitors and subsequently shown to stimulate functions in mature myeloid and other cells. GM-CSF is expressed similarly in humans and mice and its effects are mediated by binding to cell surface receptors composed of a GM-CSF–binding α-chain (CD116) and an affinity-enhancing β-chain (CD131). Ligand binding activates intracellular signaling via multiple pathways including signal transducer and activator of transcription 5 (STAT5) regulating diverse functions of myeloid cells including survival, differentiation, proliferation, and priming of specific host defense functions.8,9 GM-CSF also has poorly understood effects of alveolar epithelium. In primary PAP, pathogenesis is caused by disruption of GM-CSF signaling by neutralizing GM-CSF autoantibodies in autoimmune PAP or by recessive mutations in CSF2RA or CSF2RB (encoding GM-CSF receptor α-chain [CD116] or β-chain [CD131], respectively) in hereditary PAP.1,2,10–13
 SURFACTANT HOMEOSTASIS
SURFACTANT HOMEOSTASIS
Surfactant is vital to lung function and acts at the air–liquid–tissue interface to prevent alveolar wall collapse. It is composed of 90% lipids (largely phospholipids), 10% proteins (surfactant protein [SP]-A, -B, -C and -D), and less than 1% carbohydrate.14 SP-B and SP-C are hydrophobic phosphoproteins that contribute significantly to the surface active properties of surfactant.15 SP-A and SP-D are hydrophilic collectin family members that contribute to lung host defense.16 Surfactant lipids and proteins are synthesized, stored, and secreted into the alveoli by type II alveolar epithelial cells. In the extracellular space, surfactant “large aggregates” contribute to formation of a film that lowers surface tension and stabilizes the alveolus.17 Surfactant is expelled from the film as “small aggregates” that are internalized by type II cells and alveolar macrophages in roughly equal amounts (Fig. 70-1). Type II cells recycle and catabolize internalized surfactant equally via mechanisms that are poorly understood but do not appear to involve regulation by GM-CSF.17–19 In contrast, alveolar macrophages exclusively catabolize internalized surfactant under the positive regulatory control of GM-CSF.20,21
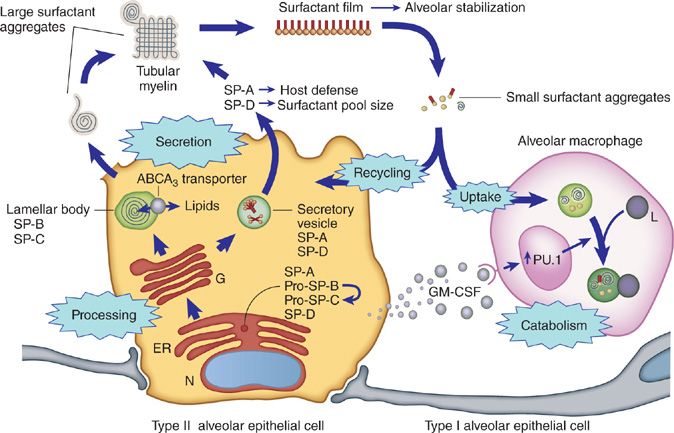
Figure 70-1 Schematic illustration depicting mechanisms of surfactant production, recycling, and catabolism. Surfactant phospholipids and proteins are synthesized in type II alveolar epithelial cells that line pulmonary alveoli. Surfactant B and C precursor proteins are processed, transported to lamellar bodies, and then secreted into the alveolar space where they interact with surfactant protein A to form tubular myelin. Surfactant monolayers and multilayers are formed from tubular myelin and function to reduce surface tension at the air–liquid–tissue interface, thus stabilizing the alveoli. Surfactant remnants are taken up and either catabolized or reutilized by type II alveolar epithelial cells. Alveolar macrophages play a critical role in surfactant homeostasis by taking up and catabolizing surfactant remnants. GM-CSF is required to maintain surfactant homeostasis and acts by stimulating catabolism of surfactant lipids and proteins in alveolar macrophages. (Reproduced with permission from Whitsett JA, Wert SE, Trapnell BC. Genetic disorders influencing lung formation and function at birth. Hum Mol Genet. 2004;13[Spec No 2]:R207–R215.)
 ANIMAL MODELS OF PAP
ANIMAL MODELS OF PAP
The discovery that GM-CSF knockout (GM-CSFKO) mice develop a pulmonary disease indistinguishable from primary PAP provided the first real pathogenic clue (Fig. 70-2).22,23 In these mice, neither production of surfactant by type II cells, nor its uptake by alveolar macrophages was impaired.22,24 Rather, surfactant clearance by alveolar macrophages was impaired.25 Replacement of GM-CSF in the lungs by direct instillation or expression of its cDNA in airway epithelium corrected the lung disease.26–28 Disruption of the GM-CSF receptor β gene (i.e., Csf2rbKO mice) caused a similar lung phenotype29 that could be corrected by bone marrow transplantation, which indicated the critical cell type driving pathogenesis was myeloid (i.e., alveolar macrophages) not epithelial (i.e., type II cells) in origin.30 Surfactant clearance in GM-CSFKO alveolar macrophages could be corrected by retroviral expression of PU.1, a transcription factor normally expressed in murine alveolar macrophages in vivo under tight regulatory control of pulmonary GM-CSF.31 These studies established that GM-CSF, via PU.1, was required for surfactant clearance in alveolar macrophages (Fig. 70-3).20 Recently, passive immunization with human PAP patient–derived neutralizing GM-CSF autoantibodies resulted in recapitulation of the cardinal pathological features of autoimmune PAP in nonhuman primates.32,33 The abnormalities induced in alveolar macrophage included impaired GM-CSF signaling, GM-CSF–dependent gene expression (including PU.1), and surfactant clearance and increased neutral lipid accumulation resulting in foamy appearing alveolar macrophages. These animal models provide strong support for disruption of GM-CSF signaling as the critical driver of PAP pathogenesis. As a model of secondary PAP, depletion of alveolar macrophages has been shown to increase lung surfactant pool size in rats.34 Congenital PAP models have been created in mice by disruption of the genes required for normal surfactant production (Sftpb, Sftpc, Abca3).35–37
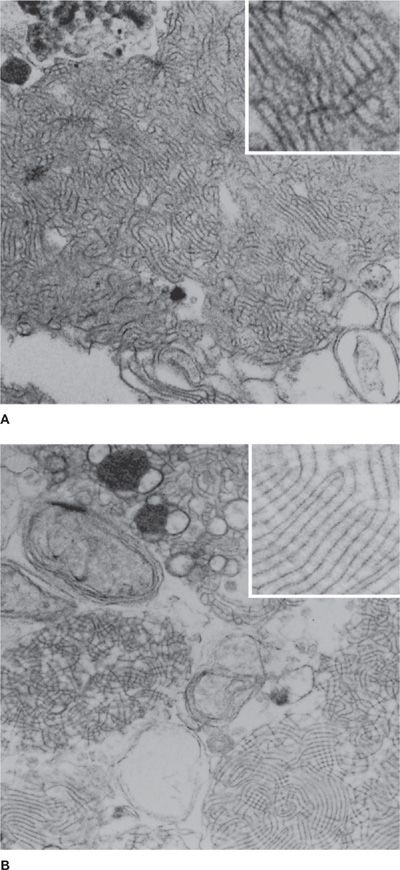
Figure 70-2 Ultrastructural appearance of the sediment from the lungs of a human patient with primary PAP (A) and a GM-CSF–deficient mouse (B). Note the presence of lamellated, fused membrane structures and amorphous debris (uranyl acetate, ×30,000).
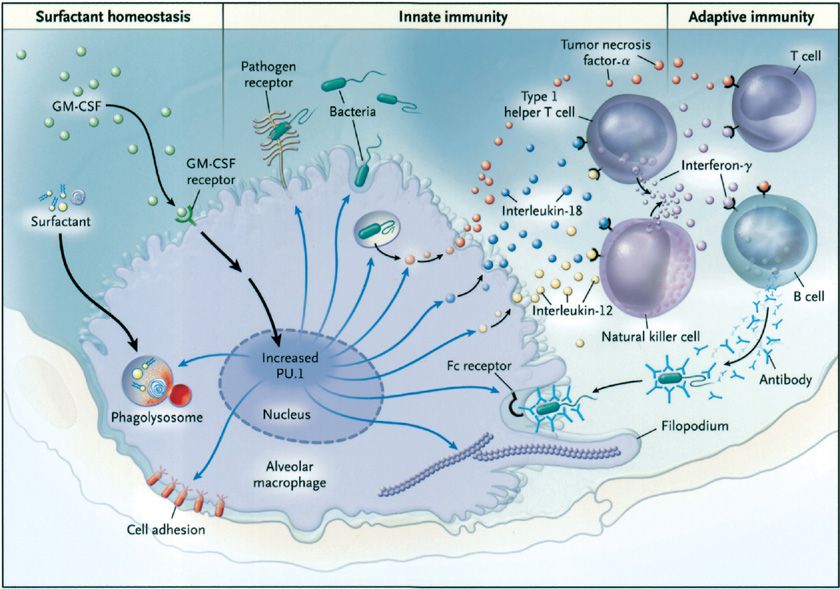
Figure 70-3 Role of GM-CSF in modulating the function of alveolar macrophages in mice. Pulmonary GM-CSF stimulates increased levels of the transcription factor PU.1 in alveolar macrophages in the lungs in vivo. Alveolar macrophages from mice deficient in GM-CSF have a number of functional defects including defects in cellular adhesion, catabolism of surfactant proteins and surfactant lipids, expression of pathogen-associated molecular pattern receptors (e.g., toll-like receptors and the mannose receptor), toll-like–receptor signaling, phagocytosis of pathogens, intracellular killing of bacteria (independent of uptake), pathogen-stimulated secretion of cytokines (tumor necrosis factor-α, interleukin [IL]-12, and IL-18), and Fc-receptor–mediated phagocytosis. Cytoskeletal organization is abnormal and may in part account for defects in phagocytosis. The ability of alveolar macrophages to release IL-12 and IL-18 severely impairs the interferon-γ response to pulmonary infection, thus impairing an important molecular connection between innate and adaptive immunity in the lung. Retroviral-mediated expression of PU.1 in alveolar macrophages from GM-CSF knockout mice corrects all these defects, suggesting that GM-CSF stimulates terminal differentiation of the macrophages primarily through the master transcription factor PU.1. The blue arrows represent the functions regulated by PU.1 that are affected by the absence of GM-CSF. (Reproduced with permission from Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 2003;349(26):2527–2539.)
 PRIMARY PAP
PRIMARY PAP
Autoimmune and hereditary forms of primary PAP are considered below.
Autoimmune PAP
In 1999, the Nakata group discovered that idiopathic PAP (also reported as acquired PAP) was specifically associated with high levels of neutralizing anti–GM-CSF autoantibodies (Fig. 70-4).10 Subsequently, these autoantibodies were shown to be (1) polyclonal, primarily comprised of immunoglobulin G subclass 1 (IgG1) and IgG2 with smaller amounts of IgG3 and IgG4, (2) have a very high affinity for GM-CSF in the 3 to 20 pM range, and (3) capable of neutralizing GM-CSF, and (4) eliminating GM-CSF bioactivity in vivo.38,39 Notwithstanding, several findings were difficult to reconcile. GM-CSF autoantibodies were also present in healthy individuals, albeit at lower levels,39 and the antibody levels in PAP patients did not correlate with disease severity.40 The recapitulation of PAP in healthy nonhuman primates by passive immunization with PAP patient–derived GM-CSF autoantibodies firmly established their critical role in pathogenesis.32,33 The low autoantibody levels in healthy individuals and lack of correlation with disease severity in PAP patients was explained by a model in which constitutive in vivo stimulation of GM-CSF–dependent myeloid cell functions (including surfactant clearance) declined with increasing GM-CSF autoantibody level until a critical threshold was reached where function became zero.39,41 The threshold level was identified and similar in humans and nonhuman primates.33,39 These studies provide strong evidence that GM-CSF autoantibodies were pathogenic and led to a recommendation to change the name from “idiopathic PAP” to “autoimmune PAP.”41
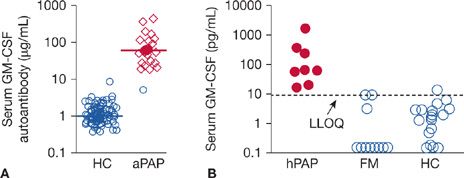
Figure 70-4 Biomarkers of use in identifying individuals with primary PAP. A. Serum GM-CSF autoantibody concentration. Shown are data for healthy controls (HCs) and patients with aPAP (autoimmune PAP). (Reproduced with permission from Uchida K, Nakata K, Suzuki T, et al. GM-CSF autoantibodies and myeloid cell immune functions in healthy individuals. Blood. 2009;113(11):2547–2556.) B. Serum GM-CSF concentration. Shown are data for children with hereditary PAP (hPAP) caused by recessive CSF2RA or CSF2RB mutations, members of their immediate family who were health (FM), or unrelated HCs. The lower limit of quantification (LLOQ) of the assay was 7.8 pg/mL (dashed line). The serum levels of GM-CSF (median, interquartile range) in children with hPAP (52 pg/mL [28–101 pg/mL]) were increased compared with healthy family members (0.0 pg/mL [0.0–3 pg/mL]) and unrelated health controls (0.0 pg/mL [0.0–1.9 pg/mL]) (n = 8, 11, 30, respectively; P < 0.001; Kruskal–Wallis analysis of variance on ranks with comparisons using Dunn’s method). (Reproduced with permission from Suzuki T, Sakagami T, Young LR, et al. Hereditary pulmonary alveolar proteinosis: pathogenesis, presentation, diagnosis, and therapy. Am J Crit Care Med. 2010;182(10):1292–1304.)
Hereditary PAP
While humans with GM-CSF deficiency have not been identified to date, in 2008, a GM-CSF autoantibody-negative child was found to have compound heterozygous mutations in CSF2RA (encoding GM-CSF receptor α) as the cause of familial PAP.11 Additional patients were identified by exploiting use of a biomarker, increased serum GM-CSF, to screen sera from a repository including patients with PAP of unknown etiology, which identified a cohort of patients with PAP-causing CSF2RA mutations (Fig. 70-4).42 Subsequently, CSF2RB mutations were identified as a cause of hereditary PAP.12,43 Detailed characterization were done including molecular cloning and expression studies to recapitulate the receptor dysfunction established that the pathogenesis of hereditary PAP is caused by recessive or compound heterozygous mutations in the CSF2RA or CSF2RB genes.11,12,42,43 A variety of mutation types were identified as the cause of hereditary PAP including missense mutations, nonsense mutations, small insertions and deletions, exon deletion, and gene deletion.2 Together, these studies defined the clinical presentation, pathogenesis, diagnosis, and therapy of hereditary PAP.
 SECONDARY PAP
SECONDARY PAP
Results from a national registry recently confirmed that secondary PAP occurs in association with a heterogeneous group of underlying diseases including hematological disorders, primarily myelodysplasia (76%–88% of cases), infectious diseases (2%–3%), other autoimmune diseases (7%), immunosuppression after organ transplantation (7%), and in nonhematological malignancies (5%).44 Secondary PAP also occurs in association with (and presumably due to) heavy inhalation exposure to inorganic dusts (e.g., silica, titanium, aluminum) and other gasses and fumes, or as a consequence of systemic infections, for example, during human immunodeficiency virus (HIV) infection.45 Secondary PAP appears to be caused by a reduction in the numbers or clearance capacity of alveolar macrophages, consistent with macrophage depletion studies in rats.34
 CONGENITAL PAP
CONGENITAL PAP
PAP occurs in neonates, infants, and children in association with various defects in the genes encoding SP-B, SP-C, ABCA3 – a lipid transporter expressed in type II alveolar epithelial cells, or TTF-1 – a transcription factor essential for lung development and surfactant expression.4,46–49 In contrast to primary and secondary PAP that are caused by reduced surfactant clearance, these disorders result from production of abnormal surfactant. While surfactant accumulation does occur to varying degrees in surfactant production disorders, these disorders are clinically, histopathologically, and pathogenically distinct from primary and secondary PAP (see below).
 THE EMERGING ROLE OF GM-CSF IN INNATE IMMUNITY
THE EMERGING ROLE OF GM-CSF IN INNATE IMMUNITY
GM-CSFKO mice have increased susceptibility to a broad range of microbial pathogens, increased mortality from spontaneous infections,50,51 and numerous host defense defects in macrophages and neutrophils.31,52 In macrophages, all these diverse defects could be corrected by forced expression of PU.1 (Fig. 70-3).31 Similar defects are present in alveolar macrophages and neutrophils from humans and nonhuman primates with disruption of GM-CSF signaling.2 GM-CSFKO mice also have systemic immune and inflammatory abnormalities51 including reduced severity in arthritis models,53
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


