Fig. 20.1
Cases of pulmonary alveolar microlithiasis in the international literature: subdivision by continent (656 pts. out of 670)
Table 20.1
Cases of pulmonary alveolar microlithiasis from 56 countries
Algeria (5) | India (50) | Peru (9) |
Argentina(2) | Iran (6) | Poland (11) |
Australia (2) | Iraq (7) | Portugal (1) |
Austria (5) | Israel (4) | Russia (37) |
Belgium (8) | Italy (64) | South Africa (4) |
Brazil (14) | Jamaica (3) | S Arabia (8) |
Bulgaria (19) | Japan (42) | Spain (36) |
Canada (2) | Kwait (1) | Sri-Lanka (2) |
China (8) | Lebanon (5) | Switzerland (4) |
Cyprus (1) | Libia (4) | Syria (1) |
Columbia (4) | Malaysia (1) | Tanzania (1) |
Egypt (2) | Mexico (5) | Thailand (3) |
France (32) | Morocco (6) | Tunisia (1) |
Germany (36) | Nigeria (2) | Turkey (105) |
Great Britain (8) | N Zealand (4) | USA (47) |
Greece (5) | Norway (9) | Yemen (1) |
Hungary (5) | Pakistan (3) | Ex Yugoslavia(11) |
Corea (1) | Giordania (1) | Uruguay (1) |
Sweden (1) |
Reviews on Turkish and Italian cases [7, 8] show that almost half of the cases were only published in local journals and that the true incidence of PAM has probably been underestimated. The majority of cases involved students, since the disease develops mainly in the first decades of life (Fig. 20.2); however housewives, office employees, workers etc. were also involved. PAM was described in association with other diseases, e.g. mitral stenosis, kidney stones or calcifications in the seminal vesicles or other organs [9]. Furthermore, the disease does not seem to only affect humans and it also appears to occur in some animals such as orang-tangs, dogs, sheep, cats, nakt mice, Afghan pika.
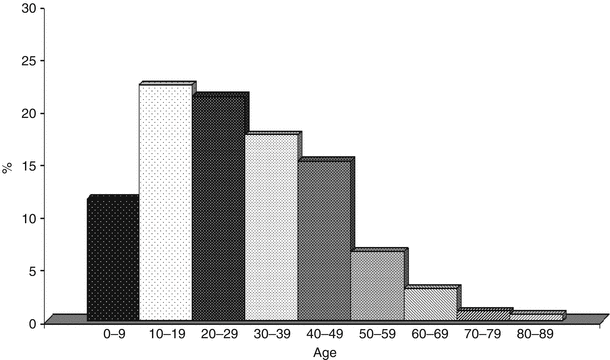
Fig. 20.2
Distribution by age of 643 pts. out of 670 cases of Pulmonary Alveolar Microlithiasis
Etiopathogenesis
Many observations support the hypothesis that a familiar inherited trait is involved according to an autosomal recessive transmission. Several cases (up to six) were diagnosed in the same family [11]. Family occurrence was found up to 31.4 % and most cases were siblings. Other authors [5, 7] found this percentage to be higher. The difference could be due to the absence of family screening for educational or economic reasons, especially in the developing countries.
In the past it was assumed that calcium salt deposits in the alveoli were due to changes in calcium metabolism because of some unknown defect. The widely endorsed hypothesis suggested an abnormality involving the carbonic anhydrase enzyme at the alveolar surface, with consequent alkalinity in the alveoli, calcium precipitation and development of calcospherites. Other hypotheses considered the first step of the disease to be the consequence of the arrival of calcified mycetes, transported into the alveoli by inhaled air; others took it to be the consequence of a diet rich in calcium salts, e.g. milk (milk alkali syndrome), or of the prolonged inhalation of dust or powdered substances (“snuff syndrome”), or changes in pulmonary haemodynamics due to mitral stenosis. Calcium metabolism was reported to be normal in most cases.
In 2006, a mutation of SLC34A2 gene was for the first time proved to occur in PAM patients [12]. This gene is a member of the solute carrier family 34 (sodium phosphate), member 2 – SLC34A2, which plays a major role in the homeostasis of inorganic phosphate [12], and it was shown by immunohistochemistry to be expressed in lungs only in alveolar type II cells, which are responsible for surfactant production [15].
The gene regulates the normal production of type IIb sodium-dependent (NaPi-IIb) phosphate transporter [16]. The function of this protein, encoded by the member 2 of SLC34A gene (SLC34A2), is to uptake liberated phosphate from the alveolar fluid for surfactant production [12]. The type II cells produce pulmonary surfactant, of which phospholipids are essential constituents; outdated surfactant is taken up by type II cells for recycling and degradation, and by alveolar macrophages only for degradation. Degraded phospholipids release phosphate that should be cleared from the alveolar space [17] (Fig. 20.3a). Dysfunction of SLC34A2 reduces the clearance of phosphate and leads to the formation of microliths composed of calcium and phosphate [13, 18] (Fig. 20.3b).
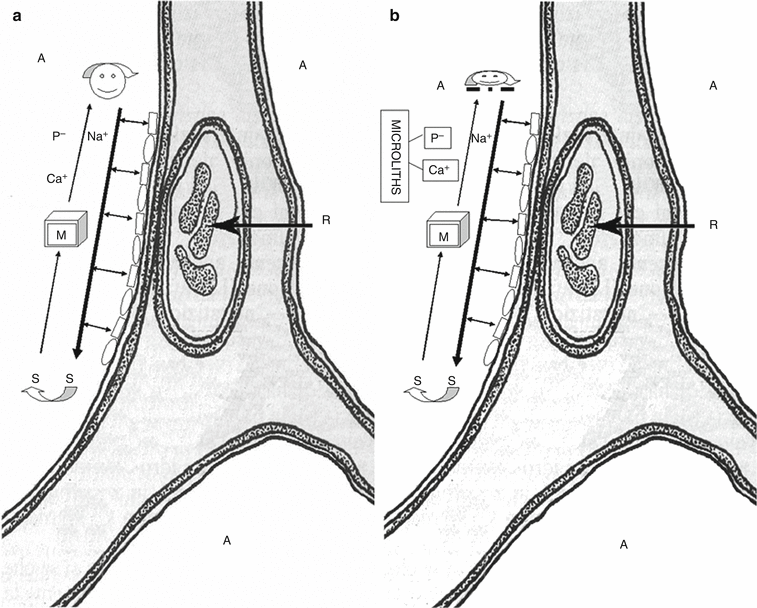
Fig. 20.3
(a) Schematic representation of the normal function of the cotransporter protein, coded by SLC34A2 gene, in the clearance of the surfactant from the alveoli. The type II pneumocytes produce the surfactant and pour it into the alveolar space. Phospholipids are essential constituents of the pulmonary surfactant and the type II cells degrade them and recycle the products of their metabolism. Na+ and P− are cleared from the alveolar space by the protein coded by SLC34A2 gene which co-transports them into the cells. (b) Schematic representation of dysfunction of the cotransporter caused by mutation of the gene SLC34A2. Dysfunction of SLC34A2 reduces the clearance of phosphate and leads to the formation of microliths composed of calcium and phosphate. A alveolar space; R red blood cell
PAM is a recessively inherited disease and is not caused by environmental factors. It has full penetrance, since none of the unaffected members is homozygous for the disease haplotype [12]. The homozygous mutations in SLC34A2 determine symptomatic PAM, while the heterozygous carriers are asymptomatic. The identification of the gene responsible for PAM facilitates genetic diagnosis and gives hope for a genetic therapy [14, 19].
Pathology
Calcium deposits within the alveoli begin at the lower lobes and in 20–30 years or more they extend to the whole lungs. Calculi have a round or ovoidal shape and range from 0.01 to 2.8 mm in diameter (Fig. 20.4). In the early stages, the alveolar septa are intact and gas exchanges are normal. Microliths progressively increase in number inside the alveoli, until they occupy the entire alveolar space and come into contact with the walls which in the advanced stages are pressed, injured and replaced by fibrous tissue. Upon opening of the chest, lungs reveal an increase in volume and weight, they are not collapsible and, upon cutting, they reveal a granular and irregular surface; the resistance of the lung parenchyma is markedly increased, almost as if it were a stone. This was precisely the picture that Malpighi [19] described in 1686 in the postmortem report of a young man – “…Pulmones turgidi graves et compactae substantiae inaequaliter nigri. Interius in vesciculis pulmonum innumeri lapilli reperti sunt …” (The lungs were heavy and compact with patches of black. Countless small stones were found in the interior of the lungs); this has also been identified as the first case of PAM reported in the literature. In all autoptic observations, the weight of the lungs is reported to be significantly increased up to 6.190 kg. Several authors have studied the structure and the composition of the microliths in an attempt to explain the pathogenesis of the disease. So it was thus observed that these concretions are irregularly concentric and laminated, and that they consist of calcium salts. Transmission or scanning electron microscopy of microlith fragments revealed spectra where calcium and phosphorus have peak intensities, and a 2:1 ratio of calcium over phosphorus, which is consistent with a Ca2PO3 content. Deposits of iron, zinc, aluminium and magnesium are frequently found in the fragments.
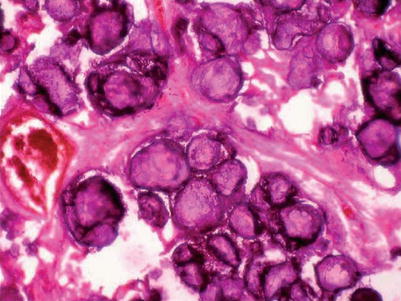
Fig. 20.4
Histopathology section showing multiple calcospherites within the alveoli of lung parenchyma. H and E 100× (Courtesy of Gayathri Devi and coll [20])
Clinical Features and Course
Symptoms and Signs
The age of PAM diagnosis has been reported to vary but it is usually diagnosed under the age of 30 years and the disease develops silently for many years. There are no symptoms in the majority of patients (up to 52.8 % of cases) and diagnosis is often fortuitous. For instance, the diagnosis has often been made during occasional chest x-rays and general check-ups required by employers, or after performing chest x-rays because of late physical or endocrine development in children [9]. Lung function tests are normal for a long time and only in the advanced stage of the disease is there an impairment, due to a restrictive syndrome or a respiratory/heart failure. Symptoms are due to an increasing number of tiny calculi within the alveoli throughout the lungs. Dyspnoea is the most frequent symptom, followed by cough, chest pain and asthenia (Fig. 20.5). It is hypothesized that coughing could be related to the direct stimulation of the C fibers or receptors by the microliths.
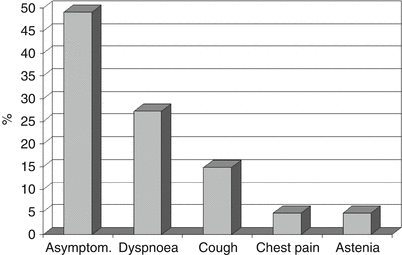
Fig. 20.5
Symptoms at diagnosis in 640 pts. out of 670 suffering from pulmonary alveolar microlithiasis
Physical examination reveals bilateral crackles and rhonchi. Their extension depends on the stage of the disease: they are limited to the bases in the early stages and extended to the intermediate or high parts of pulmonary fields in advanced stages. Sometimes cyanosis or finger clubbing are the first sign of the disease and in advanced stages cardiac-respiratory failure can worsen clinical features.
Disease Course
As mentioned above, patients may be asymptomatic for many years before appearing the respiratory failure [10]. In some cases, the disease remains apparently static; in others, over time it progresses to pulmonary fibrosis and respiratory failure accompanied by dyspnea and worsening of pulmonary function test [9]. In our experience, the radiological changes (HRCT) and functional ones (spirometry, arterial blood gas analysis) associated with the assessment of symptoms and signs were the only investigations that could assess the risk of disease progression. This is even more important when in the family there are cases of full-blown microlithiasis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


