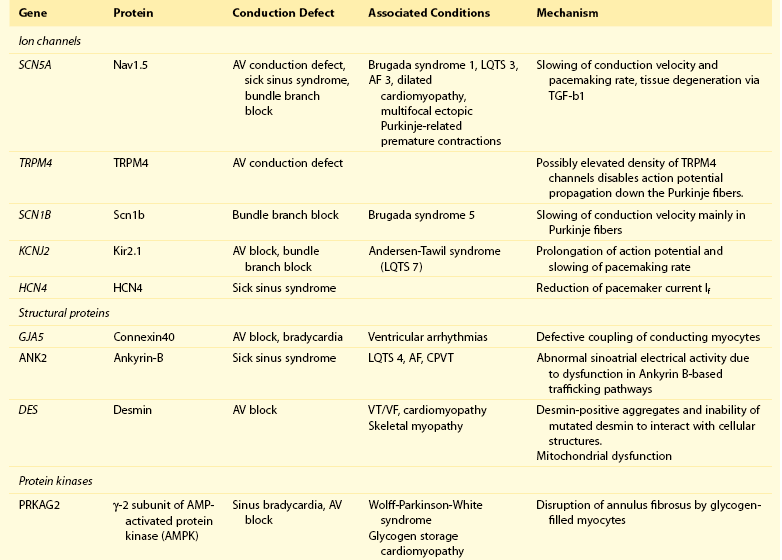
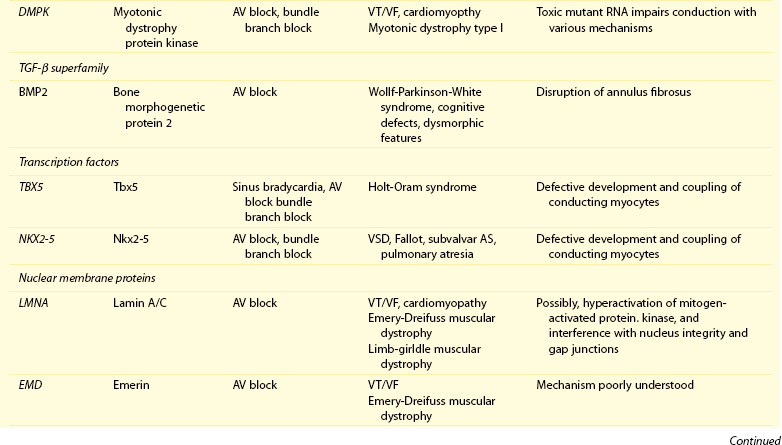
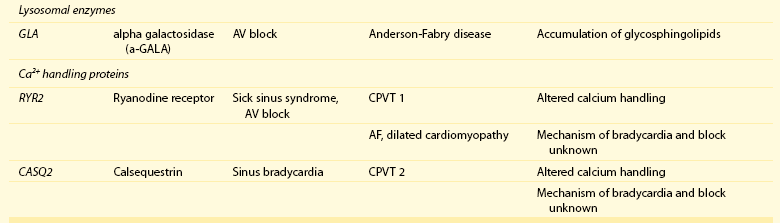
105 Progressive conduction system disease is a slow, degenerative process that affects the cardiac conduction system at any level and manifests as sinoatrial exit block, atrioventricular (AV) block at the nodal or Hisian level, or bundle branch block of varying severity. It was initially described by Lev1 and Lenègre2 as progressive AV block of unknown origin with myocardial sclerosis and loss of conduction fibers that develops over decades. Patients may present with bradycardia, a long P wave, prolonged PR and/or QRS intervals, fascicular blocks, or AV block of various types. Clinical and echocardiographic examinations are otherwise unremarkable in isolated conduction system disease. Progressive conduction disease may also be seen in the context of other disorders, with symptoms and signs of the associated condition being present (Box 105-1). Patients with prolonged QRS duration, even without bundle branch block, are at increased risk for future pacemaker implantation and may warrant monitoring for progressive conduction disease.3 Progressive myocardial degeneration with increased myocardial collagen turnover and fibrosis of the conduction system are the main histopathologic findings in patients with Lev and Lenègre disease.4,5 Lev and Lenègre hypothesized that progressive conduction defects were due to a primary degenerative disease of unknown origin that was exaggerated by aging. However, fibrosis of the conducting system may also be seen in the elderly in the absence of conduction defects.6 Evidence suggests that progressive conduction system disease has genetic and autoimmune origins (see Box 105-1).7–12 The most common causes of isolated AV conduction disease are mutations in the gene SCN5A that encodes for the pore-forming α-subunit of the cardiac sodium channel (Nav1.5) and affects depolarization phase 0 (Table 105-1 and Figure 105-1). These mutations result in slowing of conduction and fibrosis, and the cardiac conduction defect deteriorates progressively with age, leading to life-threatening conduction blocks only in the elderly.13 Thus, in the Online Mendelian Inheritance in Man database of the U.S. National Center for Biotechnology Information, progressive familial heart block (PFHB) type IA, or Lenègre-Lev disease, is classified as a disorder caused by SCN5A mutations on locus 3p21. PFHB type IB is due to mutations in the gene TRPM4 on locus 19q13.33, which encodes for the calcium-activated nonselective cation channel of the transient receptor potential melastatin. Figure 105-1 Proteins and mechanisms related to cardiac conduction system disease. Please see text and Table 105-1 for details. SCN5A as well as mutations in genes encoding for other ion channels may also result in isolated or overlapping phenotypes of sick sinus syndrome, AV conduction defects, and conditions such as the Brugada syndrome, the long QT syndrome (LQTS), dilated cardiomyopathy, and multifocal ectopic Purkinje-related premature contractions.14–23 Novel gene families associated with human conduction and affecting both PR and QRS intervals (such as SCN10A) are also being discovered.24–26 Progressive conduction disease may be seen with neuromuscular disorders, congenital cardiac defects, and Wolff-Parkinson-White syndrome secondary to mutations in genes affecting the structure or coupling of conducting myocytes via several mechanisms (see Table 105-1). Infective and autoimmune disorders may result in progressive conduction system disease caused by chronic inflammation and fibrosis, and the osteoblast phenotype of calcific aortic stenosis may involve the conduction system at any level. Both loss- and gain-of-function SCN5A mutations may slow pacemaking rates and affect conduction to surrounding atrial tissue by promoting tissue degeneration via transforming growth factor (TGF)-β1–mediated fibrosis.19,22,27,28 Cases of congenital sick sinus syndrome have been ascribed to recessive SCN5A mutations that may also lead to bradycardia in the fetus or the infant29; in adults bradycardia due to SCN5A mutations has been seen in the context of conduction disease and Brugada syndrome.16 The pacemaker or funny current (If) is generated by the hyperpolarization-activated cyclic nucleotide–gated (HCN) channel. Familial sinus bradycardia has been linked to HCN4 mutations,30 but clinical cases are usually benign with intact chronotropic competence and do not necessitate pacemaker implantation.31 Sinus bradycardia has been noted in other genetic syndromes as well, such as catecholaminergic polymorphic ventricular tachycardia. In the 1960s, Lev noticed progressive AV left bundle branch block (LBBB) in the elderly population,1 and Lenègre reported defects in the middle and distal portions of both bundle branches in relatively younger patients.2 Familial investigation of patients with bifascicular or complete AV block suggested that genetic factors could be involved in the pathogenesis of the disease,32–35 and in 1995, a locus for progressive familial heart block was mapped to chromosome 19q13.2-13.3.36,37 Subsequently, loss-of-function mutations of the cardiac sodium channel SNC5A in chromosome 3p2138,39 and gain-of-function mutations of the gene TRPM4 at chromosomal locus 19q13.340,41 were identified. Specific loss-of-function SCN5A mutations demonstrate varied competing gating effects that reduce sodium current density and enhance slow inactivation with selective slowing of conduction velocity, in a way that results in isolated conduction system disease without provoking tachyarrhythmias.39,42 Histologic examination of the heart of patients with SCN5A mutations has revealed severe degenerative changes in the conduction system,43 and Nav1.5 deficiency was found to cause tissue degeneration via TGF-β1–mediated fibrosis.22 Thus, SCN5A mutations may exert additional effects in cellular processes beyond their electrophysiological properties. SNC5A mutations cause most cases of isolated conduction disease and are usually autosomal dominant but with reduced penetrance and variable expressivity. Initial electrophysiological studies in mutation carriers with AV block revealed prolonged HV but normal AH intervals, thus suggesting infra-Hisian block, but progressive AV block due to SCN5A mutations can be both supra- and infra-Hisian.7 The TRPM4 gene encodes the transient receptor potential cation channel, subfamily M, member 4 (melastatin). A gain-of-function mutation of the TRPM4 channel that is highly expressed in cardiac Purkinje fibers has also been associated with isolated progressive cardiac conduction block.40,41 Described families with TRPM4 mutations have conduction blocks transmitted with an autosomal dominant inheritance and incomplete penetrance. Usually these patients present with right bundle branch block (RBBB) and prominent R waves in right precordial leads.41 These mutations may account for a significant portion of inherited forms of RBBB (25%) or AV block (10%).44 Sodium channel β1-subunit (SCN1B) mutations that have been associated with Brugada syndrome may also cause phenotypes of isolated progressive conduction disturbances mainly by affecting Purkinje fibers.23 SCN1B encodes for a modulatory β-subunit of the sodium channel (Scn1b) and also affects phase 0 of the action potential. Disruption of the gap junction protein connexin40, which predominantly expresses in the atria and the His-Purkinje system,45 results in prolonged AV conduction.46 A novel germ-like mutation in GJA5, the gene encoding connexin40, has been described as a cause of familial progressive heart block, slow heart rate, and malignant ventricular arrhythmias. The disease has an early onset and is associated with sudden cardiac death.47 Both loss- and gain-of-function SCN5A gene mutations may cause sick sinus syndrome, Brugada syndrome, LQTS, arrhythmia-prone dilated cardiomyopathy, and multifocal ectopic Purkinje-related premature contractions with right and/or left bundle branch block patterns.14–21,48,49 The same SCN5A gene mutation can lead to conduction disease, Brugada syndrome, or LQTS 3, although LQTS 3 mutations induce a gain-of-function in the sodium channel, whereas conduction disease or Brugada syndrome mutations induce loss-of-function in this channel.15,48,49 Affected individuals may manifest electrocardiographic features of both syndromes with QT interval prolongation at slow heart rates and distinctive ST segment elevations with exercise.48 Functional analysis has demonstrated that hereditary conduction disease may be caused by a haploinsufficiency mechanism (i.e., the single functional copy of the gene does not produce enough protein), which, in combination with aging, leads to progressive alteration in conduction velocity and results in AV block only in elder carriers.18 Why haploinsufficiency leads to Brugada syndrome in some families and to conduction disease in others is not known. Modifier genes, allele penetrance, truncation mutations that potentially reduce the sodium current to a greater extent than missense mutations,50 and/or environmental factors and sex51 may influence both the severity and the phenotype induced by an SCN5A mutation. Most of the described mutations in the SCN5A gene are autosomal dominant, but recessive mutations have also been described. Homozygotes usually present with conduction disorders and LQTS, sick sinus syndrome, or ventricular arrhythmias.52,53 Sodium channel b1 subunit (SCN1B) mutations have been associated with conduction disturbances in the context of Brugada syndrome. Mutations were located in b1 or in the newly described transcript b1B, and coexpression of mutant b-subunits with Nav1.5 resulted in diminished sodium current.23 Mutations in KCNJ2 are found in the autosomal dominant Andersen-Tawil or long QT 7 syndrome.54 KCNJ2 encodes for the inward rectifier potassium channel (Kir2.1) and affects phases 3 and 4 (i.e., terminal repolarization and resting potential). Inactivation of the inward rectifier potassium channel results in action potential prolongation and slowing of heart rate and conduction.55 Besides QT interval prolongation, potassium-sensitive paralysis, and hypoplastic mandible, patients may present with conduction abnormalities, such as AV block, bundle branch block, or nonspecific intraventricular conduction delay.56 Mutations affecting the transcription factor 5 (Tbx5) and the homeobox factor (NKX2-5) that facilitate development of the conduction lineage57 result in defective expression of gap junction connexin proteins such as Cx40 and incomplete development of the entire AV conduction system.58–60 Mutations in Tbx5 cause Holt-Oram syndrome.61 Patients with Holt-Oram syndrome manifest variable degrees of progressive conduction system dysfunction, such as sinus bradycardia and atrioventricular block, even in the absence of atrial septal defect (ASD) or other overt structural heart disease. Mutations in the gene encoding factor NKX2-5 cause nonsyndromic, congenital heart disease, mainly secundum atrial septal defects, as well as tetralogy of Fallot, ventricular septal defects (VSDs), pulmonary atresia, and subvalvular aortic stenosis and hypoplasia of the conduction system.62 These mutations are associated with progressive AV node dysfunction and other diffuse conduction system abnormalities. Absence of heart block in other major structural heart defects, as well as the presence of AV block in the absence of other malformations in mutation carriers, suggests that both NKX2-5 and TBx5 have specific roles in conduction system development and integrity, independent of their roles in structural heart development. In a small percentage of cases, WPW syndrome is familial and is associated with cardiac hypertrophy albeit without the characteristic features of hypertrophic cardiomyopathy. A missense mutation in the gene PRKAG2 that encodes the regulatory γ-subunit of adenosine monophosphate (AMP)-activated protein kinase (AMPK) has been identified.63 Patients with mutations on the PRKAG2 gene have a variable combination of glycogen storage cardiomyopathy, progressive conduction system disease including sinus bradycardia and atrioventricular block, ventricular preexcitation, arrhythmias, and sudden death.64,65 The R302Q mutation in PRKAG2 has been associated with Mahaim fibers.66 AV block in patients with PRKAG2 mutations is progressive and infra-Hisian. The annulus fibrosus, which normally insulates the ventricles from inappropriate excitation by the atria, is thinned and disrupted by glycogen-filled myocytes. These anomalous microscopic atrioventricular connections, rather than morphologically distinct bypass tracts, appear to provide the anatomical substrate for ventricular preexcitation. A novel form of WPW syndrome is associated with microdeletion in the region of gene BMP2, which encodes the bone morphogenetic protein-2, a member of the TGF-β gene superfamily, and affects the development of annulus fibrosus. It is characterized by variable cognitive deficits, dysmorphic features, and prolonged AV conduction on atrial pacing.67
Progressive Conduction System Disease
Definition
Clinical Presentation
Pathophysiology
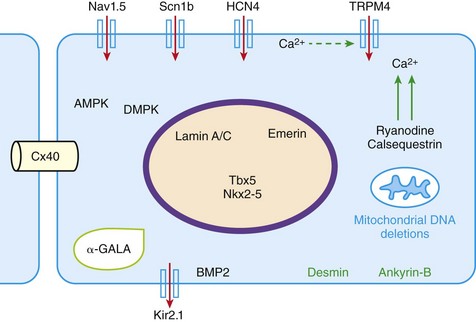
Clinical Entities
Familial Sick Sinus Syndrome
Idiopathic Isolated Conduction Disease (Progressive Familial Heart Block Types IA and IB)
Conduction Disease in the Context of Genetic Arrhythmia Syndromes
Congenital Heart Deformities
Wolff-Parkinson-White (WPW) Syndrome and Cardiomyopathy
Progressive Conduction System Disease