PRINCIPLES OF ELECTROPHYSIOLOGY
David D. Spragg ![]() Gordon F. Tomaselli
Gordon F. Tomaselli
HISTORY AND INTRODUCTION
The field of cardiac electrophysiology was ushered in with the development of the electrocardiogram (ECG) by Einthoven at the turn of the twentieth century. The recording of cellular membrane currents revealed that the body surface ECG is the timed sum of the cellular action potentials in the atria and ventricles. In the late 1960s, the development of intracavitary recording, in particular the ability to record His bundle electrograms with programmed stimulation of the heart, marked the beginning of contemporary clinical electrophysiology. Adoption of radio frequency technology to ablate cardiac tissue in the early 1990s heralded the birth of interventional cardiac electrophysiology.
The clinical problem of sudden death caused by ventricular arrhythmias, most commonly in the setting of coronary obstruction, was recognized as early as the late nineteenth century. The problem was vexing and led to the development of pharmacologic and nonpharmacologic therapies, including transthoracic defibrillators, cardiac massage, and, most recently, implantable defibrillators. Over time the limitations of antiarrhythmic drug therapy have been highlighted repeatedly in clinical trials, and now ablation and devices are first-line therapy for a number of cardiac arrhythmias.
In the last two decades, the genetic basis of a number of heritable arrhythmias has been elucidated, revealing important insights into the mechanisms not only of these rare arrhythmias but also of similar rhythm disturbances observed in more common forms of heart disease.
DESCRIPTIVE PHYSIOLOGY
The normal cardiac impulse is generated by pacemaker cells in the sinoatrial node situated at the junction of the right atrium and the superior vena cava (see Fig. 11-1). This impulse is transmitted slowly through nodal tissue to the anatomically complex atria, where it is conducted more rapidly to the atrioventricular node (AVN), inscribing the P wave of the ECG (see Fig. 11-2). There is a perceptible delay in conduction through the anatomically and functionally heterogeneous AVN. The time needed for activation of the atria and the AVN delay is represented as the PR interval of the ECG. The AVN is the only electrical connection between the atria and the ventricles in the normal heart. The electrical impulse emerges from the AVN and is transmitted to the His-Purkinje system, specifically the common bundle of His, then the left and right bundle branches, and then to the Purkinje network, facilitating activation of ventricular muscle. In normal circumstances, the ventricles are activated rapidly in a well-defined fashion that is determined by the course of the Purkinje network, and this inscribes the QRS complex (see Fig. 11-2). Recovery of electrical excitability occurs more slowly and is governed by the time of activation and duration of regional action potentials. The relative brevity of epicardial action potentials in the ventricle results in repolarization that occurs first on the epicardial surface and then proceeds to the endocardium, which inscribes a T wave normally of the same polarity as the QRS complex. The duration of activation and recovery is determined by the action potential duration represented on the body surface ECG by the QT interval (see Fig. 11-2).
Cardiac myocytes exhibit a characteristically long action potential (200–400 ms) compared with neurons and skeletal muscle cells (1–5 ms). The action potential profile is sculpted by the orchestrated activity of multiple distinctive time- and voltage-dependent ionic currents (Fig. 14-1A). The currents in turn are carried by transmembrane proteins that passively conduct ions down their electrochemical gradients through selective pores (ion channels), actively transport ions against their electrochemical gradient (pumps, transporters), or electrogenically exchange ionic species (exchangers).
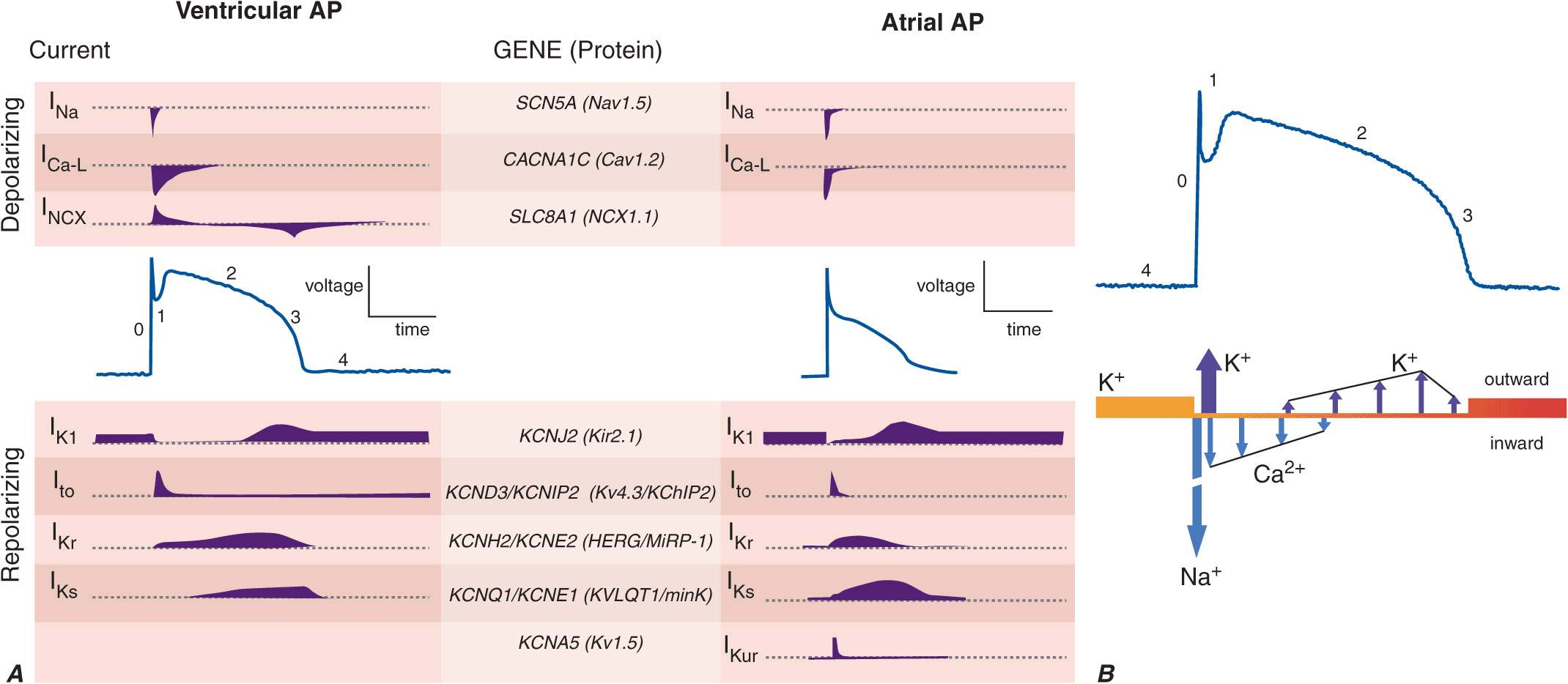
FIGURE 14-1
A. Cellular atrial and ventricular action potentials. Phases 0–4 are the rapid upstroke, early repolarization, plateau, late repolarization, and diastole, respectively. The ionic currents and their respective genes are shown above and below the action potentials. The currents that underlie the action potentials vary in atrial and ventricular myocytes. B. A ventricular action potential with a schematic of the ionic currents flowing during the phases of the action potential. Potassium current (IK1) is the principal current during phase 4 and determines the resting membrane potential of the myocyte. Sodium current generates the upstroke of the action potential (phase 0); activation of Ito with inactivation of the Na current inscribes early repolarization (phase 1). The plateau (phase 2) is generated by a balance of repolarizing potassium currents and depolarizing calcium current. Inactivation of the calcium current with persistent activation of potassium currents (predominantly IKr and IKs) causes phase 3 repolarization.
Action potentials in the heart are regionally distinct. The regional variability in cardiac action potentials is a result of differences in the number and types of ion channel proteins expressed by different cell types in the heart. Further, unique sets of ionic currents are active in pacemaking and muscle cells, and the relative contributions of these currents may vary in the same cell type in different regions of the heart (Fig. 14-1A).
Ion channels are complex, multisubunit transmembrane glycoproteins that open and close in response to a number of biologic stimuli, including a change in membrane voltage, ligand binding (directly to the channel or to a G protein–coupled receptor), and mechanical deformation (Fig. 14-2). Other ion motive exchangers and transporters contribute importantly to cellular excitability in the heart. Ion pumps establish and maintain the ionic gradients across the cell membrane that serve as the driving force for current flow through ion channels. Transporters or exchangers that do not move ions in an electrically neutral manner (e.g., the sodium-calcium exchanger transports three Na+ for one Ca2+) are termed electrogenic and contribute directly to the action potential profile.
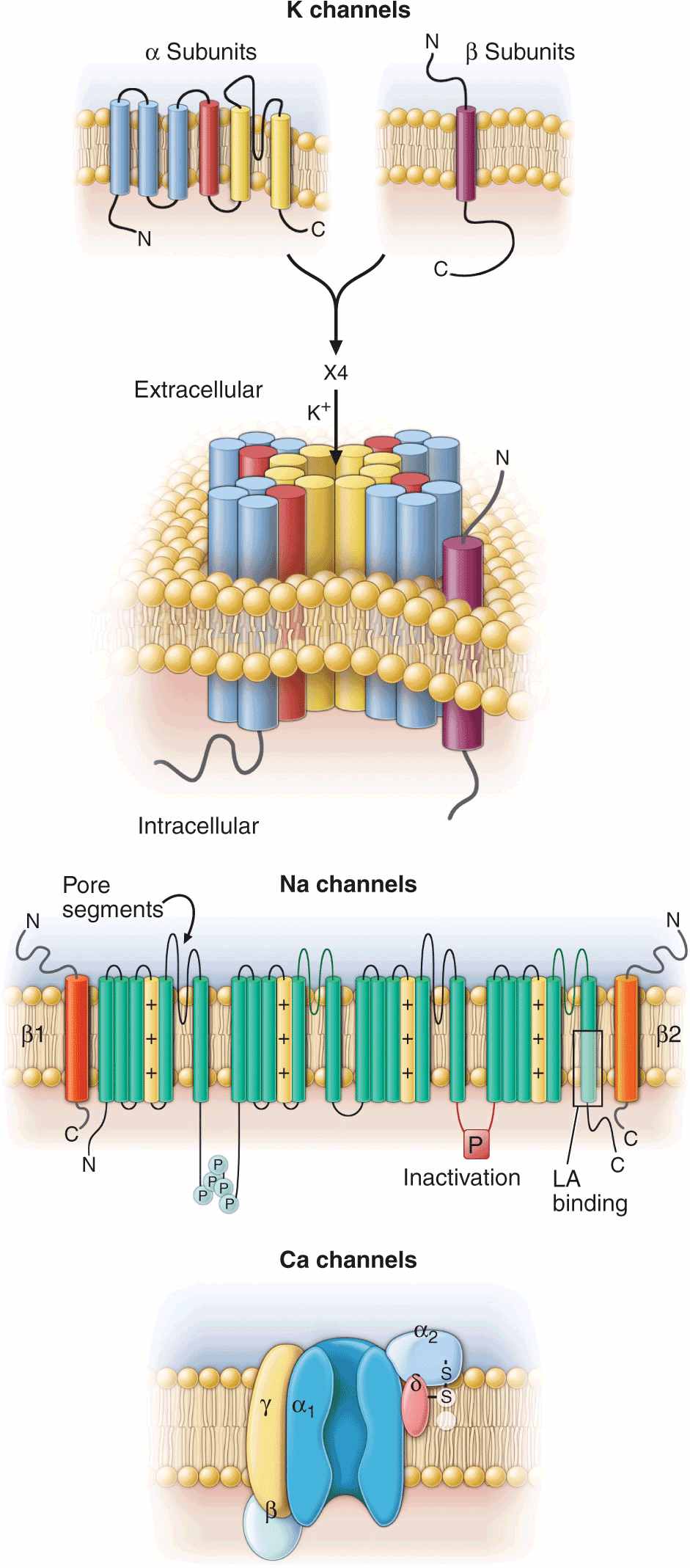
FIGURE 14-2
Topology and subunit composition of the voltage-dependent ion channels. Potassium channels are formed by the tetramerization of α or pore-forming subunits and one or more β subunits; only single β subunits are shown for clarity. Sodium and calcium channels are composed of α subunits with four homologous domains and one or more ancillary subunits. In all channel types the loop of protein between the fifth and sixth membrane-spanning repeat in each subunit or domain forms the ion-selective pore. In the case of the sodium channel, the channel is a target for phosphorylation, the linker between the third and fourth homologous domain is critical to inactivation, and the sixth membrane-spanning repeat in the fourth domain is important in local anesthetic antiarrhythmic drug binding.
The most abundant superfamily of ion channels expressed in the heart is voltage gated. Several structural themes are common to all voltage-dependent ion channels. First, the architecture is modular, consisting either of four homologous subunits (e.g., K channels) or of four internally homologous domains (e.g., Na and Ca channels). Second, the proteins fold around a central pore lined by amino acids that exhibit exquisite conservation within a given channel family of like selectivity (e.g., jellyfish, eel, fruit fly, and human Na channels have very similar P segments). Third, the general strategy for activation gating (opening and closing in response to changes in membrane voltage) is highly conserved: the fourth trans-membrane segment (S4), studded with positively charged residues, lies within the membrane field and moves in response to depolarization, opening the channel. Fourth, most ion channel complexes include not only the pore-forming proteins (α subunits) but also auxiliary subunits (e.g., β subunits) that modify channel function (Fig. 14-2).
Na and Ca channels are the primary carriers of depolarizing current in both the atria and the ventricles; inactivation of these currents and activation of repolarizing K currents hyperpolarize the heart cells, reestablishing the negative resting membrane potential (Fig. 14-1B). The plateau phase is a time when little current is flowing, and relatively minor changes in depolarizing or repolarizing currents can have profound effects on the shape and duration of the action profile. Mutations in subunits of these channel proteins produce arrhythmogenic alterations in the action potentials that cause long and short QT syndrome, idiopathic ventricular fibrillation, familial atrial fibrillation, and some forms of conduction system disease.
MECHANISMS OF CARDIAC ARRHYTHMIAS
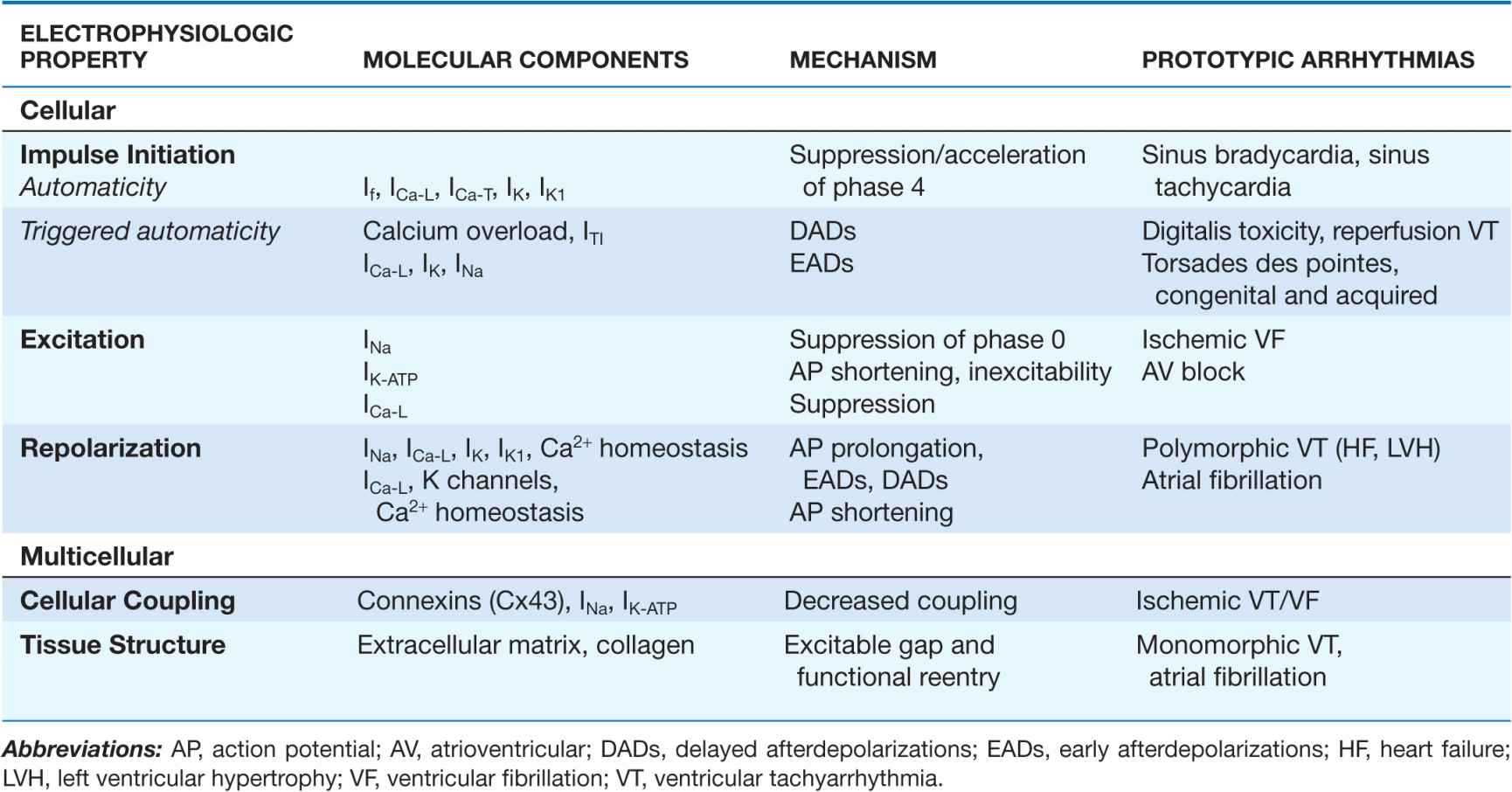
Cardiac arrhythmias result from abnormalities of electrical impulse generation, conduction, or both. Bradyarrhythmias typically arise from disturbances in impulse formation at the level of the sinoatrial node or from disturbances in impulse propagation at any level, including exit block from the sinus node, conduction block in the AV node, and impaired conduction in the His-Purkinje system. Tachyarrhythmias can be classified according to mechanism, including enhanced automaticity (spontaneous depolarization of atrial, junctional, or ventricular pacemakers), reentry (circus propagation of a depolarizing wavefront), or triggered arrhythmias (initiated by afterdepolarizations) occurring during or immediately after cardiac repolarization, during phase 3 or 4 of the action potential. A variety of mapping and pacing maneuvers typically performed during invasive electrophysiologic testing can often determine the underlying mechanism of a tachyarrhythmia (Table 14-1).
TABLE 14-1
ARRHYTHMIA MECHANISMS
Alterations in impulse initiation: Automaticity
Spontaneous (phase 4) diastolic depolarization underlies the property of automaticity (pacemaking) characteristic of cells in the sinoatrial (SA) and atrioventricular (AV) nodes, His-Purkinje system, coronary sinus, and pulmonary veins. Phase 4 depolarization results from the concerted action of a number of ionic currents, including K+ currents, Ca2+ currents, electrogenic Na, K-ATPase, the Na-Ca exchanger, and the so-called funny, or pacemaker, current (If); however, the relative importance of these currents remains controversial.
The rate of phase 4 depolarization and, therefore, the firing rates of pacemaker cells are dynamically regulated. Prominent among the factors that modulate phase 4 is autonomic nervous system tone. The negative chronotropic effect of activation of the parasympathetic nervous system is a result of the release of acetylcholine that binds to muscarinic receptors, releasing G protein βγ subunits that activate a potassium current (IKACh) in nodal and atrial cells. The resulting increase in K+ conductance opposes membrane depolarization, slowing the rate of rise of phase 4 of the action potential. Conversely, augmentation of sympathetic nervous system tone increases myocardial catecholamine concentrations, which activate both α– and β–adrenergic receptors. The effect of β1-adrenergic stimulation predominates in pacemaking cells, augmenting both L-type Ca current (ICa-L) and If, thus increasing the slope of phase 4. Enhanced sympathetic nervous system activity can dramatically increase the rate of firing of SA nodal cells, producing sinus tachycardia with rates >200 beats/min. By contrast, the increased rate of firing of Purkinje cells is more limited, rarely producing ventricular tachyarrhythmias >120 beats/min.
Normal automaticity may be affected by a number other factors associated with heart disease. Hypokalemia and ischemia may reduce the activity of Na, K-ATPase, thereby reducing the background repolarizing current and enhancing phase 4 diastolic depolarization. The end result would be an increase in the spontaneous firing rate of pacemaking cells. Modest increases in extracellular potassium may render the maximum diastolic potential more positive, thereby also increasing the firing rate of pacemaking cells. A more significant increase in [K+]o, however, renders the heart inexcitable by depolarizing the membrane potential.
Normal or enhanced automaticity of subsidiary latent pacemakers produces escape rhythms in the setting of failure of more dominant pacemakers. Suppression of a pacemaker cell by a faster rhythm leads to an increased intracellular Na+ load ([Na+]i), and extrusion of Na+ from the cell by Na, K-ATPase produces an increased background repolarizing current that slows phase 4 diastolic depolarization. At slower rates, [Na+]i is decreased, as is the activity of the Na, K-ATPase, resulting in progressively more rapid diastolic depolarization and warm-up of the tachycardia rate. Overdrive suppression and warm-up are characteristic of, but may not be observed in, all automatic tachycardias. Abnormal conduction into tissue with enhanced automaticity (entrance block) may blunt or eliminate the phenomena of overdrive suppression and warm-up of automatic tissue.
Abnormal automaticity may underlie atrial tachycardia, accelerated idioventricular rhythms, and ventricular tachycardia, particularly that associated with ischemia and reperfusion. It has also been suggested that injury currents at the borders of ischemic myocardium may depolarize adjacent nonischemic tissue, predisposing to automatic ventricular tachycardia.
Afterdepolarizations and triggered automaticity
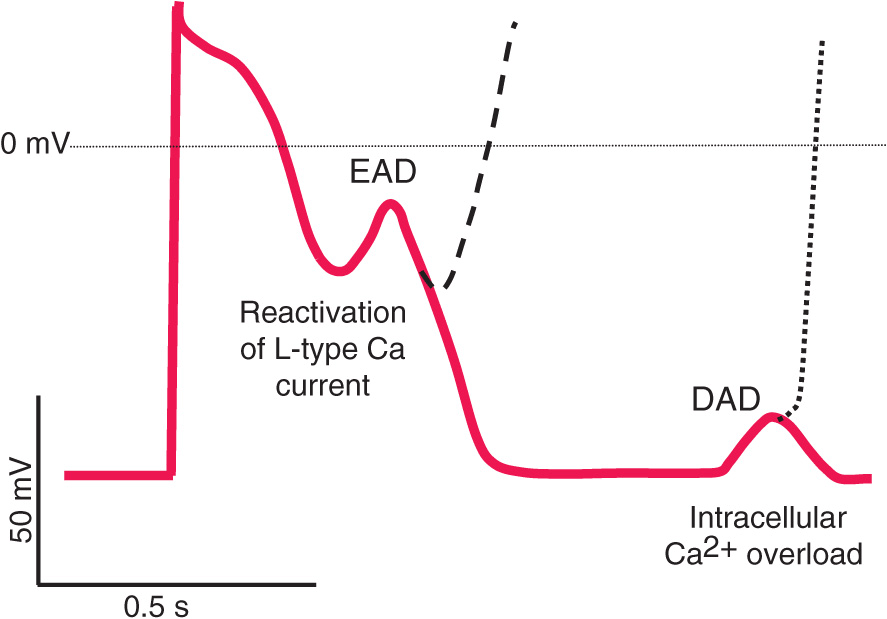
Triggered automaticity or activity refers to impulse initiation that is dependent on afterdepolarizations (Fig. 14-3). Afterdepolarizations are membrane voltage oscillations that occur during (early afterdepolarizations, EADs) or after (delayed afterdepolarizations, DADs) an action potential.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


