Chapter 52 Portal Hypertension
Pathogenesis
Physiologically, portal hypertension results from either an increase in the portal blood flow (rare) or an obstruction to the outflow of blood from the portal circulation (common). Obstructions to the portal circulation have been classified anatomically based on their location relative to the hepatic sinusoids. Accordingly, the obstruction may be presinusoidal, sinusoidal, or postsinusoidal. Presinusoidal and postsinusoidal obstructions have been subclassified as intrahepatic or extrahepatic (Box 52-1).
Intrahepatic Presinusoidal Obstruction
Schistosomiasis is the most common cause of portal hypertension in third-world countries. Deposition of ova in the portal vein walls results in a granulomatous inflammatory reaction, which in turn results in fibrosis and portal flow restriction. Hepatic function is preserved in the early stages, but later stages of this disease are characterized by advanced cirrhosis and loss of hepatic function.1 Myeloproliferative disorders such as myelosclerosis and myeloid leukemia occasionally lead to presinusoidal hypertension by virtue of the deposition of primitive cellular material infiltrating the portal zones.2 Sarcoidosis causes portal hypertension by two mechanisms: sarcoid granulomas within the portal vein leading to obstruction, and increased portal blood flow.
Intrahepatic Sinusoidal and Postsinusoidal Obstruction
Two mechanisms account for the portal hypertension in these patients. First is the mechanical obstruction of the portal blood flow by the regenerating hepatic nodules and cirrhotic bands within the damaged liver. These changes may extend beyond the confines of the hepatic sinusoids, accounting for the presence of presinusoidal, sinusoidal, and postsinusoidal distortion of the hepatic architecture. The second element is an increase in the splanchnic perfusion, in part attributed to the genesis of multiple arteriovenous shunts and collateral channels. One third of portal blood flow may bypass functional hepatocytes through these channels.3 The clinical correlate of this increased blood flow is the hyperdynamic state that typifies cirrhosis: elevated cardiac output and a diminished systemic resistance.4
Extrahepatic Postsinusoidal Obstruction
Postsinusoidal hepatic vein obstruction is usually the result of thrombosis in the hepatic veins. Although the cause of most cases is unknown, a number of associated diseases have been identified. Membranous webs of the hepatic veins, malignancies (hepatomas, renal carcinomas, adrenal carcinomas), trauma, pregnancy, contraceptive use, acute alcoholic hepatitis, veno-occlusive disease, and Senecio species (ragwort) toxicity may all result in hepatic vein thrombosis.5 Constrictive pericarditis and chronic congestive heart failure may also cause postsinusoidal obstruction.
Budd-Chiari syndrome is the result of hepatic venous occlusive disease and is characterized by massive ascites, esophageal varices, variceal hemorrhage, hepatic failure, and death. Chiari disease is due to primary hepatic vein ostial occlusion. The clinical progression after hepatic vein occlusion may be fulminant or gradual. Hepatic failure is the result of chronic congestion and ischemia from impaired hepatic blood flow. The factors that determine the rate of progression are not well understood. Angiography is essential in establishing the diagnosis; it identifies the presence of thrombus and its location.6,7
An initial trial of anticoagulation may allow endogenous fibrinolysis to resolve the venous thrombosis. Patients whose course is gradually progressive and who have intact hepatocellular function should be considered for portal decompression by a portacaval, mesocaval, or mesoatrial shunt. Shunt selection is dependent on the patient’s anatomy, as defined by angiography. When the Budd-Chiari syndrome leads to deterioration of hepatic function, as demonstrated by abnormal liver function tests, hepatic transplantation is the procedure of choice.8
Diagnosis
Hemodynamic measurement of the portal circulation is most commonly accomplished by transjugular venous catheterization and measurement of the wedge hepatic vein pressure.9 This technique is able to record the pressure in the hepatic veins and the hepatic sinusoids. Elevations of the wedge pressure reflect elevations in the portal venous pressure. False-negative results may be encountered in patients with presinusoidal obstruction and in cases of catheter malfunction. Normal hepatic venous pressure is essentially the same as the right atrial pressure (0 to 5 mm Hg); portal venous pressure is approximately 2 to 6 mm Hg higher than hepatic venous pressure. A gradient greater than 10 mm Hg is considered abnormal.
Other methods of measuring portal venous pressure have been developed but have largely been abandoned because of the increased risk associated with them. These tests include direct cannulation of the portal vein (requires surgical exposure), percutaneous transhepatic portal venous catheterization, transjugular portal vein catheterization, and percutaneous splenic pulp pressure measurement.10
The most recent development in assessing the portal circulation is the application of noninvasive ultrasonography. Duplex scanning has been used to establish patency and measure the direction and velocity of portal blood flow. Information gathered from this technique identifies portal vein thrombosis, hepatopetal and hepatofugal flow, and portal hypertension.9–12 Bolondi and associates13 documented high sensitivity and specificity with these techniques.
Diagnostic Evaluation
Laboratory Testing
The initial step in evaluating most patients is to assess their serum chemistries. Specific attention should be placed on testing the liver enzymes (serum glutamic-oxaloacetic transaminase [SGOT], serum glutamic-pyruvic transaminase [SGPT], lactate dehydrogenase [LDH], alkaline phosphatase) and hepatic synthetic function (prothrombin time and serum albumin). Information from these two sets of tests identifies patients who are suffering from acute hepatocellular damage and those who have had sufficient damage to reduce the liver’s ability to synthesize essential proteins. This represents two distinct gradations of hepatic dysfunction. The first is indicative of an acute insult; the second represents the degree of hepatic dysfunction. The presence and degree of abnormalities in liver function tests correlate with outcome: the more abnormal the test results, the worse the prognosis.14,15
Abnormal liver function tests combined with physical findings and historical data form the basis for classifying patients with portal hypertension. The Child classification or, more recently, the combined Child-Pugh classification serves as a prognosticator of survival in cirrhotic patients who undergo both emergent and elective surgery (Table 52-1).16
Additional laboratory investigations should include a determination of serum ammonia and a complete blood count (CBC)—white blood cells (WBCs), red blood cells (RBCs), and platelets. Serum ammonia may be elevated in cases of severe hepatic dysfunction and coma. It correlates loosely with mentation but may serve as an indicator of a treatable cause of encephalopathy, hyperammonemia.17
The CBC detects the presence of anemia and hypersplenism. Anemia in cirrhotic patients can result from a number of causes other than hemorrhage. Chronic malnutrition is a particularly important cause of anemia in these patients. Although splenomegaly is present in virtually all portal hypertensive patients, hypersplenism may not develop until later in the course of the disease. The size of the spleen does not correlate directly with either the degree of portal hypertension or the severity of hypersplenism, but an enlarged spleen is found in virtually all patients with portal hypertension and hypersplenism.18 Hypersplenism is defined principally in terms of splenic sequestration and destruction of platelets and WBCs. This leads to significant depressions in the platelet and WBC counts. Platelet counts less than 50,000/mm3 and WBC counts less than 2000/mm3 support this diagnosis.
Upper Gastrointestinal Endoscopy
The diagnosis of portal hypertension can be established by noting the presence of varices. The size, appearance, and location of the varices may significantly affect the patient’s management. Endoscopy also notes the presence of other sources of bleeding in portal hypertensive patients, such as hypertensive gastropathy, gastritis, gastric ulceration, duodenal ulceration, gastric mucosal lacerations (Mallory-Weiss tears), or esophageal ulcerations. Because of the variety of possible bleeding lesions and the significant differences in the management of these lesions, patients admitted for hemorrhage must undergo upper gastrointestinal endoscopy on each admission. As many as 40% to 60% of patients with documented varices have associated gastritis or peptic ulcer disease.19 In patients with esophageal and gastric varices, the gastric varices have been documented as the site of bleeding in up to 18%.20
Liver Biopsy
Mikkelsen and others noted an operative mortality of 69% in elective shunt cases and 83% in emergent cases in the presence of acute hyaline necrosis.20,21 Other authors have contested whether acute alcoholic hepatitis alters survival.14,22 Finally, it should be noted that Mallory bodies disappear when patients abstain from alcohol and the liver recovers from the insult.23
Duplex Scanning
Duplex scanning is finding greater application in the evaluation of portal hypertensive patients. In patients who are being considered for portacaval shunting or hepatic transplantation, the duplex scan is frequently sufficient to document portal vein patency. Duplex scanning determines both the patency of the portal vein and the direction of portal venous blood flow. This is the minimal anatomic information required to proceed with these operations. The combination of color-flow imaging and duplex scanning has improved the accuracy and extended the diagnostic abilities of duplex scanners.11
Angiography
Preoperative anatomic definition is essential for optimal surgical management, particularly when peripheral shunts are being considered. If possible, angiography should be performed on all patients who are to undergo elective shunting procedures. Techniques that are of primarily historical interest include splenoportography24 (introduction of radiopaque material into the spleen), umbilical vein catheterization, and transhepatic percutaneous portal venography.21–30
The goal of these studies is to delineate the major portal tributaries—the splenic vein, the superior mesenteric vein (SMV), and the portal vein itself—and their relation to the renal vein. An additional goal of the liver package is to measure the hepatic wedge pressure and visualize the hepatic sinusoidal circulation. These last two elements are helpful in confirming the presence of portal hypertension, estimating the severity of the hypertension, and determining the cause of the elevated pressure. Low hepatic wedge pressure (<10 to 12 mm Hg) in a patient with variceal hemorrhage should prompt a careful search for evidence of portal vein thrombosis.31 The wedge hepatic vein catheter allows determination of the morphology of the sinusoids and the direction of blood flow (Figure 52-1). The wedge hepatic venogram in cirrhotic patients demonstrates irregular sinusoids with multiple scattered filling defects. Retrograde portal vein filling indicates hepatofugal flow.24
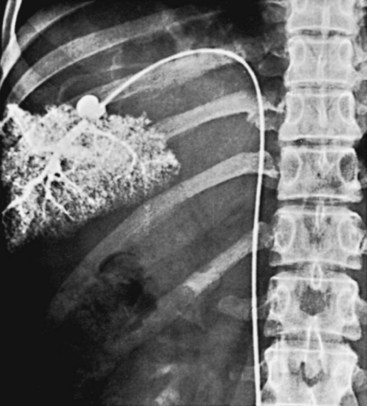
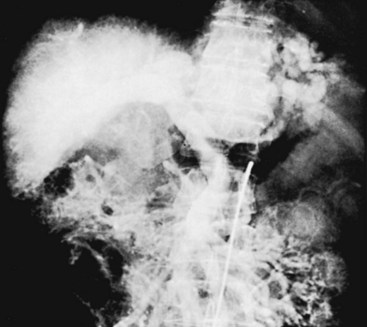
Delineation of the portal tributary anatomy is essential in planning an elective portal decompressive procedure, because the choice of procedure is limited by the patient’s anatomy. The angiographic findings correlate with the degree of cirrhosis. In early cirrhosis, no definite angiographic abnormalities are present. As cirrhosis becomes more severe, one sees the development of collateral pathways, dilatation of the hepatic artery, and pruning of intrahepatic portal vein branches (Figure 52-2). In advanced cirrhosis, reversal of flow in the portal vein may be detected.
Complications
Esophageal Varices
Any blood vessel that is attenuated and distended under supranormal pressures is at risk of disruption and bleeding. The addition of mechanical trauma or chemical irritation may increase the likelihood of bleeding. Consequently, any of the collaterals that develop because of portal hypertension may bleed. Hemorrhoidal vessels, intestinal varices, and stomal varices have all been documented as bleeding sites in cirrhotic patients. The mechanical and chemical irritants that bathe the gastroesophageal region result in esophagitis, attenuation of the mucosal layers, and disruption of the varices. When the increased blood pressure within the varices is combined with periodic exacerbations of this pressure by activities that increase the intraabdominal and intrathoracic pressure (e.g., coughing, retching), the risk of bleeding from esophageal varices is significantly increased. The elevation of portal pressure results in dilatation of all these collateral pathways (Figure 52-3).
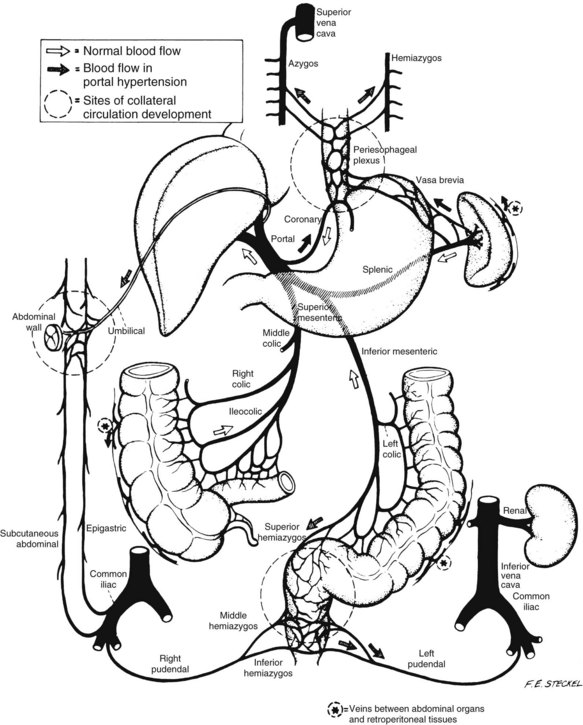
FIGURE 52-3 Schematic diagram of collateral venous pathways.
(From Sedgwick CE, et al: Portal hypertension, Boston Little Brown, 1967.)
Several attempts have been made to predict the risk of hemorrhage from esophageal varices. Characterization of the severity of portal hypertension on the basis of corrected sinusoidal pressure has not correlated with subsequent hemorrhage. Factors that predict the risk of bleeding include the size of the varices, the Child class of the patient, and the presence of erosions on the varices (red-dot signs).32–36
Encephalopathy
Although not usually considered a life-threatening complication of hepatic failure, encephalopathy can have a profoundly disabling effect on patients. The clinical manifestations of encephalopathy are varied and cover a spectrum from mild inattention to frank coma. The most commonly used system of staging encephalopathy classifies patients from stage I through stage IV. Progression begins with mild personality alterations and occasionally with asterixis or clonus in stage I. Stage II may be characterized by drowsiness, sometimes with mild confusion. Stage III is typified by stupor and obtundation. Coma is the hallmark of stage IV. Electroencephalography is not specifically diagnostic, characteristically showing only slow wave activity, primarily in the frontal regions.37
Elevated ammonia levels have several significant repercussions. First, they elevate glucagon levels. This in turn stimulates gluconeogenesis, which produces more ammonia. In addition, the gluconeogenesis leads to elevated insulin levels, which promotes catabolism of branched chain amino acids. This ultimately leads to increased levels of straight chain amino acids such as phenylalanine, tyrosine, and methionine. An elevated ratio of straight chain to branched chain amino acids drives neutral amino acids past the blood-brain barrier. The cerebral uptake of these neutral amino acids is possible because ammonia stimulates brain glutamine synthesis, allowing rapid equilibration of brain glutamine for straight chain neutral amino acids. These same neutral amino acids may act as false neurotransmitters and are thought to produce encephalopathy.38
The treatment of encephalopathy is based on reduction of ammonia levels and supplementation of branched chain amino acids. Lactulose and neomycin reduce ammonia uptake from the gut by altering the intestinal pH, reducing the number of intestinal bacteria, and reducing intestinal transit of protein. Other agents such as levodopa have been used, with mixed results, in improving encephalopathy.39
Medical Therapy
Management of Acute Complications
The management of acute complications is a highly specialized area in the care of these patients. The most common problem that requires urgent care is hemorrhage. The significance of hemorrhage in these patients is difficult to understate. Hemorrhage may be associated with up to 70% mortality, depending on the cause and severity of the liver disease and the degree of decompensation of the patient at the time of presentation. Furthermore, the risk of a second hemorrhage within 1 year may be as high as 60%.16
The essential steps in caring for a cirrhotic patient with an upper gastrointestinal hemorrhage include establishing peripheral venous access, volume-resuscitating the patient, and determining the source of bleeding. This is best accomplished by upper gastrointestinal endoscopy. As mentioned previously, between 40% and 60% of cirrhotic patients with known varices who have an upper gastrointestinal hemorrhage are not bleeding from their varices.40
Specific Measures for the Control of Acute Hemorrhage
Protein and Gut Lavage
The upper gastrointestinal tract should be laved with the aid of a large-bore tube (e.g., Ewald tube) to remove the clotted blood from the stomach. The rest of the tract is cleansed at the appropriate time by the administration of lactulose and neomycin by mouth. Neomycin and lactulose are given to alter the intestinal absorption of ammonia. Neomycin is a relatively nonabsorbable antibiotic; it destroys urease-producing bacteria and thereby decreases ammonia production. Lactulose is converted by lactase-containing intestinal bacteria into lactic acid and acetic acid, thus decreasing the intraluminal intestinal pH. The lower pH ionizes ammonia into ammonium (NH4+), which is less able to diffuse through the colonic mucosa. Lactulose also promotes diarrhea, cleansing the intestine of its contents.17,41,42
Vasopressin
Vasopressin (Pitressin) should no longer be considered first-line therapy in the management of active bleeding varices. However, information from its use has provided significant insights into the early management of variceal bleeding. The drug is specifically directed at slowing and stopping variceal hemorrhage. It has been recognized since 1917 for its vasoconstrictive effects and its ability to decrease portal pressure.43 Vasopressin is a naturally occurring nonapeptide that demonstrates general vasoconstrictive effects, with particular efficacy in the splanchnic bed. This splanchnic vasoconstriction leads to decreased portal flow. Vasopressin is also known to diminish cardiac output by an average of 14% and heart rate by 11%. This, in turn, reduces hepatic blood flow by approximately 44% and wedge hepatic vein pressure by 11%. As much as a 23% reduction in the gradient between hepatic venous pressure and wedge hepatic vein pressure has been documented.44
A consequence of the vasoconstrictive effects of vasopressin is the potential exacerbation of cardiac ischemia. To counter these ischemic effects, a number of pharmacologic agents have been used in conjunction with vasopressin.44–46 Isoproterenol, when administered with intravenous vasopressin, results in an equivalent reduction of portal vein pressure but maintenance of cardiac output.45 Sublingual nitroglycerin plus vasopressin has been shown to reduce the deleterious effects of vasopressin alone while preserving the decrease in portal vein pressure.44,47 In the course of a controlled trial, Gimson and associates48 noted that nitroglycerin may reverse some of the cardiac suppressive effects of vasopressin, as well as enhance the portal hypotensive effects.
Somatostatin and Octreotide
Because of its fewer side effects and equal efficacy compared with vasopressin, octreotide has become the agent of choice in managing acute variceal hemorrhage. Somatostatin is a tetradecapeptide derived from the hypothalamus that has demonstrated an ability to decrease splanchnic blood. Octreotide is a synthetic octapeptide analog of somatostatin. It has a longer half-life than somatostatin (100 minutes vs. 2 to 3 minutes). Both have been demonstrated to be as effective as vasopressin for the control of acute bleeding, with fewer complications.49 Hemodynamic studies indicate that somatostatin decreases the portal venous pressure gradient.42,50 Randomized studies have shown equal efficacy between the two, and metaanalysis has demonstrated no survival benefit. Somatostatin has been repeatedly shown to have fewer side effects than vasopressin.49,51
Octreotide is effective in reducing and halting variceal hemorrhage in 80% of cases. Intravenous octreotide should be used before, and along with, balloon tamponade. It should be started before endoscopy. The initial dose of octreotide is a bolus infusion of 50 µg. This is followed by an infusion of 50 µg/h. As with vasopressin infusions, the octreotide infusion should be continued over a 3-day period. After this period, the drug should be gradually discontinued.46
Data have been mixed regarding somatostatin and related agents, but several studies demonstrated decreased blood loss when these drugs are used. For example, a metaanalysis comprising 12 trials (1452 patients) showed an overall decrease in transfusion requirements of 1 unit of blood per patient.52 Mortality, rebleeding, and need for balloon tamponade were not decreased. As the authors concluded, the transfusion requirements are significant, but it is not evident that there is any clinical benefit in saving a single unit of blood.
Terlipressin is a synthetic vasopressin analog that can be given intermittently as an injection, unlike vasopressin, which must be administered as an intravenous drip. In comparison with vasopressin, no significant differences in outcome were shown in a metaanalysis of 20 studies comprising 1609 patients.53 It is not clear whether this drug will become a front-line treatment agent. In summary, because these drugs are easy to administer, have relatively safe profiles, and may be effective, many centers use them routinely for bleeding varices. Additional support for their use comes from a recent update of a Cochrane Database review demonstrating that vasoactive drugs appeared to work well when sclerotherapy was not readily available. The therapy was also found to be associated with few risks.54
Propranolol
Evidence of its efficacy in the reduction of recurrent bleeding has been mixed. Burroughs and colleagues55 compared propranolol with placebo and found no significant difference in rebleeding or survival. Fleig and colleagues56 randomized 70 patients to sclerotherapy or propranolol and found no difference in the rebleeding rate or survival; however, propranolol decreased the size of the varices significantly after 3 months of treatment. In a prospective, controlled, randomized study of 79 patients, Lebrec and associates57,58 noted that after 3 months, 2.6% of patients maintained on propranolol had recurrent gastrointestinal tract bleeding, compared with 66% maintained on a placebo drug. Similar findings have been reported by others.59 Poynard and coauthors analyzed 127 patients treated with propranolol and found five factors that were associated with rebleeding: (1) hepatocellular carcinoma, (2) lack of persistent decrease in heart rate, (3) lack of abstinence from alcohol, (4) lack of compliance, and (5) prior history of rebleeding60 More recently, Schepke and associates61 compared propranolol therapy to band ligation in primary prophylaxis of varices in high-risk patients, noting that there were no differences in bleeding between these treatments. More recent data suggest that sclerotherapy is more effective in this regard, reducing the risk of a first variceal bleeding episode to 5% versus a cohort receiving propranolol alone, which had a risk of 20%.62
Balloon Tamponade
All these tubes work on the same principle, tamponade of varices. The design variations include the presence of one or two balloons for compression of the stomach alone or the esophagus and stomach (Linton-Nachlas vs. Sengstaken tube) and the presence of adjunctive ports for aspiration of the stomach and esophageal secretions (Sengstaken-Blakemore vs. Edlich modification or Minnesota tube).63,64 The Sengstaken-Blakemore tube is probably used more often because it can compress both esophageal and gastric varices, whereas the Linton-Nachlas tube can compress only gastric varices.
Once the gastric balloon is inflated, it is taped to the facemask of a football helmet with approximately 1 kg of pressure. The gastric and esophageal ports are connected to intermittent low Gomco suction. The position of the gastric balloon is checked periodically to ensure that migration into the esophagus has not occurred. If bleeding does not cease with gastric balloon inflation and tension, the esophageal balloon is inflated to 24 to 45 mm Hg pressure. If bleeding ceases with the Sengstaken-Blakemore tube insertion, it is left inflated for 24 hours. After this period, the esophageal balloon should be deflated. Twenty-four hours later, the gastric balloon is deflated. If bleeding does not recur, the tube is deflated and left in place. It should be removed after an additional day.63 Esophageal variceal tamponade results in cessation of hemorrhage in 45% to 92% of cases.65–68 Bleeding recurs shortly after the Sengstaken-Blakemore tube is deflated in 24% to 42% of cases, however, and cannot be controlled in 33% to 37% of cases.65,66 The incidence of recurrent bleeding after a second period of balloon control is 40%.67
This tube has been associated with significant complications: gastroesophageal tears, ulceration, and perforation. Pulmonary complications include aspiration pneumonia and asphyxia from tracheal intubation. Conn and Simpson reported a complication rate of 41% and a mortality rate of 20%.69 More commonly, the incidence of major complications is in the range of 4% to 9%.65–67
Surgical Shunt Correction
Prophylactic Shunting
Proponents of prophylactic shunting argued that variceal hemorrhage could be prevented by creating a shunt in patients with varices before they had the opportunity to bleed. Four prospective, controlled studies addressed this issue.19,21,70,71 These four early studies were similar in design. The patients were divided between medical and surgical treatment and were followed for recurrent bleeding, development of encephalopathy, and survival.
The Boston Interhospital Liver Group (BILG) allocated 45 patients to the medical group and 48 to the surgical group. At 1-, 3-, and 5-year intervals, there was no difference in survival; at 5 years, the survival rate was 50%. Encephalopathy was likewise equal (21%).72 The Department of Veterans Affairs (VA) study demonstrated a 45% incidence of encephalopathy after shunting, almost twice that of medical therapies. It was disconcerting that the 5-year survival after shunting (51%) was less than with medical therapy (64%).19 Similarly, the experience of the Yale group demonstrated decreased survival after shunting, with an increased incidence of encephalopathy.73,74
Indications for prophylactic variceal decompression have also been studied by the Cooperative Study Group of Portal Hypertension in Japan. By comparing only nondecompressive transection procedures and selective shunts, this group found no difference in survival rates at 2 years and suggested that in certain patients, prophylactic procedures may be indicated.21 It should, however, be noted that these patients were primarily nonalcoholic, and the applicability of these results has been widely debated in the United States.
Therapeutic Shunting
The efficacy of portosystemic shunting has been studied with prospective, randomized clinical trials. Four such trials were performed in the United States and France to evaluate the fundamental question of whether therapeutic shunts prolong survival and maintain the quality of life compared with conventional medical therapy.75–78
The VA study, begun in 1961, followed the survival of 155 selected patients over a 5.5-year period. Although 78 patients were randomized to the surgical group, only 67 actually received shunts. Operative mortality following therapeutic portacaval shunts was 8%. Recurrence of variceal bleeding was 7% in the surgical group and 65% in the medical group. Encephalopathy occurred with approximately equal frequency in both groups, but was more severe in the shunted group. The long-term survival rate at 5 years was 57% in the shunt group and 36% in the medical group. The increase in long-term survival was not, however, statistically significant.70
In the BILG study,72 patients underwent end-to-side portacaval shunt, side-to-side portacaval shunt, or medical therapy. The long-term survival was better in the shunt group than in the medically treated group, but this difference was not statistically significant. On the basis of both the VA and the BILG studies, Conn concluded that, despite the lack of statistical significance, therapeutic portacaval shunts prolong the mean duration of life in cirrhotic patients who have suffered from variceal hemorrhage.79
Researchers at the University of Southern California published a 12-year follow-up of a prospective, randomized study comparing end-to-side portacaval shunts with medical therapy. There were 190 episodes of bleeding in the group receiving medical therapy, compared with 11 in the surgical group. Encephalopathy of a moderate to severe degree occurred in 35% of shunt patients. A 5-year life-table analysis revealed a 44% survival rate among patients treated with shunts and a 24% survival rate among those treated medically. This difference was not statistically significant, however.21
Rueff and associates, in a study at the Hôpital Beaujon in Clichy, France, compared therapeutic end-to-side portacaval shunts with medical therapy.78 The long-term survival was 47% in the shunt group and 56% in the medical group. The diminished long-term survival in the shunt group is unique to this study and may be due to the high operative mortality (19%, compared with 13% for the BILG study and 8% for the VA study). Encephalopathy was equally common in the medical and surgical groups, with an incidence of 40%. As in the other studies, it tended to be more severe and chronic in the shunt group, however. Recurrent bleeding occurred in 8% of the shunt group and in 72% of the medical group.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


