Chapter 55 Neointimal Hyperplasia
The development of strategies designed to suppress neointimal hyperplasia after peripheral vascular interventions has become increasingly important in light of the current enthusiasm for less invasive but more locally injurious percutaneous therapies. Nearly all vascular interventions are prone to the development of neointimal hyperplasia because manipulation of the arterial wall induces some form of endothelial cell injury. This injury initiates a cascade of events that ultimately results in neointimal hyperplasia and limits the durability of these vascular interventions. Some vascular interventions are more prone to the development of neointimal hyperplasia; these include prosthetic bypass and arteriovenous grafts.1 However, percutaneous balloon angioplasty with or without stenting, endovascular atherectomy, vein bypass grafting, and endarterectomy are all susceptible to this process.2–5 Thus peripheral vascular interventions will remain temporary and palliative rather than durable and potentially curative unless effective treatment of neointimal hyperplasia can be achieved.
Neointimal hyperplasia results from a complex cascade of events that involves all three layers of the arterial wall as well as circulating blood elements. Much has been learned about this process in the past decade that has changed the way neointimal hyperplasia is viewed and has opened up new strategies for therapeutic development. The classic description of neointimal hyperplasia resulting from proliferation and migration of vascular smooth muscle cells (VSMC) from the media to the intima, along with deposition of the extracellular matrix after endothelial cell injury, no longer accurately represents the pathophysiologic changes that occur during this injury process.6,7 It is now recognized that the adventitia as well as resident and circulating stem and progenitor cells play a large role in this process. Furthermore, in addition to the well-described role of cytokines and growth factors that are secreted from platelets and leukocytes at the site of injury, reactive oxygen species and adipokine have been shown to be intimately involved in the regulation of this process. This chapter summarizes the current concepts regarding the pathology and pathophysiology of neointimal hyperplasia.
Pathology
Neointimal hyperplasia can be described as the abnormal continued proliferation of cells and connective tissue elements that occurs at sites of arterial injury. During the first decade of the 20th century, Carrel and Guthrie8 noted that “within a few days after the operation, the stitches placed in making the anastomosis became covered with a glistening substance similar in appearance to the normal endothelium.” This early description of arterial healing most likely represents the normal response of an artery to injury. Neointimal hyperplasia, in contrast, is more likely the result of an exaggeration of this response through the inability to control, or the continued stimulation of, this normal regenerative process.
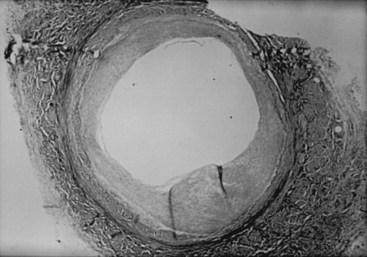
Lesions of neointimal hyperplasia are firm and homogeneous upon gross examination. They look smooth, shiny, and subendothelial. These lesions were often referred to as myointimal hyperplasia, initially highlighting the thought that the medial VSMC is the origin of the proliferating tissue. However, given that it is now recognized that multiple cell types contribute to the neointimal mass, the term neointimal hyperplasia or intimal hyperplasia is more commonly used today. Assessment of neointimal lesions using intravascular ultrasound has supported the notion that the majority (≈90%) of lesions are homogeneous in appearance. However, with advances in technology, such as intravascular optical coherence tomography, it appears that up to 40% of lesions may be heterogeneous in nature.9 Pathologic histologic examination of neointimal lesions reveals mainly stellate cells surrounded by a clear fibromyxomatous stroma and connective tissue. These stellate cells contain smooth muscle–like features and stain positively for actin and sulfated glycosaminoglycans.10,11 These appear to originate from several different sources, including the media; the adventitia (i.e., fibroblasts); the endothelium; and resident and circulating stem cells. Cellular proliferation begins within 24 hours of injury and is one of the hallmarks of neointimal hyperplasia. However, proliferation, migration, phenotypic differentiation, cellular infiltration, and extracellular matrix deposition all occur, resulting in wall thickening that continues to develop over several weeks (Figure 55-1).12
Pathophysiology
The classic description of the arterial injury response by both Clowes and Ross involves damage to the vascular endothelium and exposure of the underlying VSMC to circulating blood elements.6,7,13–15 This exposure of the subendothelial arterial wall elements triggers the activation of myriad cellular and enzymatic events. The underlying internal elastic lamina and VSMC are exposed to circulating blood elements. Platelets immediately aggregate and adhere to the site of injury.16 An inflammatory response follows, with the infiltration of neutrophils, macrophages, and leukocytes.17,18 Twenty-four hours after injury, under the influence of growth factors and cytokines, medial VSMC convert from a contractile to a synthetic phenotype and begin to proliferate.19 VSMC migrate to the neointima, where they continue to proliferate for up to 8 weeks.6,20,21 Concurrently, endothelial cell regeneration occurs through the stimulation of basic fibroblast growth factor (bFGF) within 24 hours after injury and can continue for 6 to 10 weeks.22 Finally, transforming growth factor beta (TGF-β) stimulates extracellular matrix deposition.23,24 The culmination of these events results in the formation of neointima hyperplasia (Figure 55-2).
The classic arterial injury response just described includes no mention of the adventitia. However, the adventitia is now thought to be one of the main driving forces in the development of neointimal hyperplasia. Investigators have characterized the proliferative response after arterial injury in rat and pig arteries.25,26 Common to both is that the proliferative response in the adventitia, compared with the intima and media, is much greater at almost all time points. Although 1.5-fold to twofold more proliferation has been reported in the adventitia compared with in the media in rat carotid arteries (Table 55-1), nearly sevenfold more proliferation has been reported in the adventitia than in the media in pig coronary arteries.25,26 Furthermore, upon examination of early time points, proliferation has been found to occur in the adventitia before the media at time points as early as 4 hours (see Table 55-1).
TABLE 55-1 Proliferating Cell Number/mm2 after Arterial Injury
| Time Point | Media | Adventitia |
|---|---|---|
| Noninjured | 0.2 | 0.1 |
| 4 hours | 42 | 286 |
| 8 hours | 37 | 510 |
| 12 hours | 38 | 531 |
| 24 hours | 369 | 582 |
| 48 hours | 972 | 691 |
| 72 hours | 930 (29%) | 1164 (53%) |
| 7 days | 576 (14%) | 1424 (41%) |
| 14 days | 420 (13%) | 810 (23%) |
| 30 days | 22 (0.4%) | 75 (2%) |
Modified from Couffinhal T, Dufourcq P, Jaspard B, et al: Kinetics of adventitial repair in the rat carotid model. Coron Artery Dis 12:635–648, 2001.
The development of neointimal hyperplasia in vein grafts bears a similarity to neointimal hyperplasia after arterial injury, but it differs in that there are fewer medial cells that respond and there is an additional response to increased wall tension. Prominent features in rodent models of vein grafting include extensive early denudation of the endothelium and smooth muscle cell layers, attachment of leukocytes and platelets to the subendothelial matrix, and elaboration of cytokine and growth factor products by these cells. This results in the activation, migration, and proliferation of vein graft adventitial myofibroblasts to form a neointima.27–32 Pathologically, the trauma resulting from surgical manipulation and exposure of the vein graft to arterial pressure and flow results in nearly complete denudation of the endothelium and loss of the smooth muscle cell layers within the first 24 hours (Figure 55-3). Early after denudation, attachment of leukocytes and platelets occurs. Macrophage infiltration dominates the early inflammatory changes observed with vein graft pathobiology. This prominent inflammatory response peaks during the first 2 weeks of vein graft adaptation, which exceeds the response observed in models of arterial injury (Figure 55-4). Stark and Hoch elegantly demonstrated that by inhibiting macrophage activity early in this inflammatory process, a significant reduction in neointimal hyperplasia was achieved.29,31

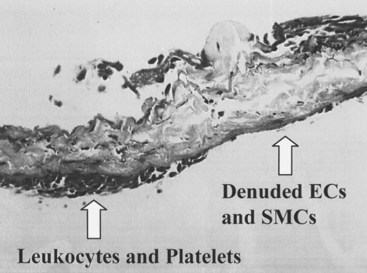
Vein graft neointimal changes persist and can progress despite re-endothelialization, in contrast to arterial neointimal proliferation, which tends to halt its progression upon restitution of the endothelial cell layer. It is believed that the ongoing neointimal thickening associated with vein graft neointimal hyperplasia is an adaptive response to arterial pressure and flow.33 Wall thickening is greater in regions of low shear (e.g., at the inner wall of a curve). Neointimal hyperplasia results in wall thickening, which decreases wall stress to more physiologic levels. These hyperplastic changes were described by Carrel and Guthrie34 when vein grafts were first examined histologically. They referred to this process as “arterialization.” It is now known that this arterialization process involved phenotypic transformation of the vein cells to an arterial phenotype, as indicated by the loss of ephrin-B4. Wall thickening can be accentuated at valve cusps, perhaps because of local flow abnormalities; at the anastomoses, where shear and wall tension are greatest; and at sites of clamp injury. Hemodynamically significant lesions caused by intimal hyperplasia usually occur after the first month and are rare after a year. After that time, vein grafts can develop lesions similar to atherosclerosis.
Hemodynamic and Mechanical Factors
A wide variety of hemodynamic and mechanical factors, including high- and low-flow velocities, high and low wall shear stress, and mechanical compliance mismatch, have been implicated in the formation of intimal hyperplasia.35–37 The effects of flow velocity on the subsequent development of intimal hyperplasia have been studied in a variety of models. In general, low blood velocity tends to favor the development of neointimal hyperplasia. For example, in a canine carotid vein interposition model, segments with low-flow velocities developed significantly thicker intimal layers38; however, there are exceptions to this rule. In a high-flow renal artery–to–vena cava anastomosis, significant intimal thickening was documented by electron microscopic examination 3 months after formation.39 Similar intimal lesions have also been noted in arteriovenous fistulas (AVF) constructed for hemodialysis access.40 This may be a result of the compliance mismatch noted between the arterial and venous systems. In a monkey iliac AVF model, 6 months after construction no increase in intimal thickening occurred on the experimental side.41 In that study, although flow rate and velocity were markedly increased on the side of the AVF, the calculated wall shear stress was equal on both sides. This equality of shear stress, despite a significant difference in flow velocities, was the result of a twofold increase in the vessel lumen diameter. The vessel wall regulates its diameter to produce a physiologic level of shear stress at the lumen. The endothelium regulates diameter by producing both vasoconstrictors such as endothelin, and vasodilators such as nitric oxide (NO). These factors also affect VSMC proliferation and when chronically present, cause remodeling of the vessel to maintain the new diameter without vasoactivity.
In other primate studies, arteriovenous flow has been shown to inhibit neointimal thickening in highly porous polytetrafluoroethylene (PTFE) grafts.42 Return to normal flow causes thickening of these lesions. In a rodent model of arterial injury, low flow caused increased intimal hyperplasia.43 These and many other studies demonstrate that intimal hyperplasia is increased by low shear and inhibited by high shear. Regions of low wall shear stress also may stimulate intimal proliferation by increasing the time for lipid transport.35,38 The results of postmortem studies have shown that early atherosclerotic lesions occur more commonly in areas of low wall shear stress.44 In the carotid artery, atherosclerosis starts at the outer wall of the bulb, where shear forces are low and oscillating.45,46
Compliance mismatch is also an important hemodynamic factor in the production of anastomotic intimal hyperplasia.47 Compliance is defined as the percentage of radial change per unit pressure and is a useful index of vessel wall distensibility to a pressure force. Although experimental and clinical studies have shown that conduits with compliance values approaching that of the native artery have better patency, none of these studies controlled for differences in graft surfaces. In one study, femoropopliteal autografts made from glutaraldehyde-treated carotid arteries had better patency when fixation produced a more compliant graft.48 However, other studies have shown that the creation of stiffer, less compliant grafts by treating arteries with glutaraldehyde results in markedly diminished patency rates.47,49 Although the compliance of a vein graft at the time of implantation is similar to that of a native artery, according to the results of one study the compliance values actually remained within the normal range for a median follow-up of 33 months.50 Textile and fabric prostheses, in contrast, are relatively noncompliant. Furthermore, 4 months after implantation, a significant loss of compliance is noted in both polyester fabric and PTFE grafts.51
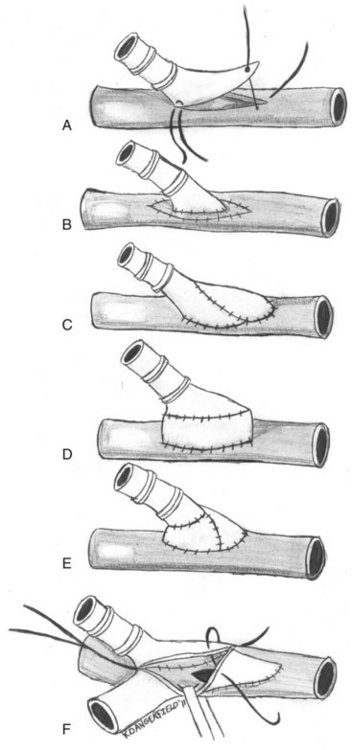
For this reason, some authors have suggested imposing a short interposition of autogenous vein between the native artery and prosthetic grafts.52 These modifications are largely intended to decrease the compliance mismatch between the prosthetic graft and the native artery. They include vein cuffs, vein patches, vein boots, and AVF at the distal anastomosis (Figure 55-5).53 A prospective randomized trial to examine the efficacy of Miller vein cuffs in improving lower extremity PTFE graft patency showed that vein cuffs did not improve patency rates in bypasses to the above-knee popliteal artery.54 However, cuffed below-knee popliteal artery grafts had a 45% 3-year patency rate versus a 19% patency rate in uncuffed grafts (p = 0.018).55 Similarly, the use of vein patches, such as those developed by Taylor and Linton, at the distal anastomosis yields better long-term patency rates versus prosthetic material alone.56,57 Although there have been no randomized, prospective trials to date, retrospective reviews have demonstrated improved patency of vein-patched PTFE versus PTFE alone when used for below-knee bypasses. Taylor and colleagues reported a 5-year primary patency of 54% for 83 infrapopliteal PTFE grafts using his vein patch modification.56 Although there is no control arm to this study, previously reported patency rates with PTFE alone are significantly worse.1 A review of 145 patients who had undergone either above- or below-knee bypasses using autologous vein, unmodified PTFE, and Linton patch–modified PTFE found that above-knee bypasses had no statistical difference in 1- and 3-year patency rates between the three groups.58 However, the cumulative patency rates for 1, 3, and 5 years after below-knee bypasses were 93%, 75%, and 75% for the autologous vein group (n = 30); 74%, 52%, and 42% for the unmodified PTFE group (n = 37); and 93%, 93%, and 93% for the Linton patch–modified PTFE group (n = 16), respectively. They speculated that the improved results over unmodified PTFE bypass grafts were a result of improved compliance mismatch between the prosthetic graft and the targeted native artery. The study was limited by the small number of Linton patch–modified PTFE grafts used compared with the autologous vein and PTFE groups. Finally, a retrospective analysis of outcomes of patients with AVF-modified PTFE grafts versus PTFE alone to the infrapopliteal arteries showed that the 2-year patency rates were significantly better for grafts with the AVF modification (23% vs. 5% for the PTFE alone grafts, p < 0.05).59 The AVF, originally described by Ascher and colleagues, is created by first mobilizing an adjacent vein and transecting it.60 The vein is then anastomosed to the artery in an end-to-side fashion. A veinotomy is created near the AVF and the ePTFE is anastomosed, creating the AVF modification at the distal anastomosis (see Figure 55-5). Despite these surgical improvements, patency rates remain poor, especially when the distal outflow is at or below the level of the popliteal artery, primarly as a result of the formation of neointimal hyperplasia.
Platelets
Platelet-derived growth factor (PDGF) is secreted along with other granule constituents, including platelet factor 4, thromboglobulin, and thrombospondin. PDGF is a cationic protein with a molecular weight of 28 to 31 kDa. It comprises two subunits (α and β). Physiologically, PDGF functions as both a chemoattractant and a mitogen for VSMC and fibroblasts. Because it binds with high affinity to VSMC, some have suggested that PDGF may attract the VSMC from the media into the intima, bind to them, and stimulate their proliferation. Interestingly, evidence has shown that the platelet may not be the only source of this protein. PDGF (α and β subunits) is produced by human umbilical vein and saphenous vein endothelial cells. In fact, a large increase in PDGF production can be measured in injured endothelial cells. Also, both α-subunit and β-subunit messenger RNA (mRNA) have been noted in fresh endarterectomy specimens obtained during carotid surgery.61 VSMC themselves also produce PDGF-like activity in response to arterial injury, and VSMC from human atheroma contain mRNA for the PDGF α subunit.62 Taken together, these findings may explain how proliferation continues after re-endothelialization occurs. PDGF may be released by both platelets and endothelial cells, causing the activation and migration of VSMC and myofibroblasts, which then secrete additional PDGF, leading to proliferation. Finally, demonstrating the importance of PDGF to the arterial injury response, inhibition of PDGF using antibodies or antisense oligonucleotides in animal models of arterial injury has been shown to inhibit neointimal hyperplasia by 60% to 80%.63–65
Inflammatory Cells
Inflammatory cells are intimately involved in the arterial response to injury. Electron microscopic studies after balloon catheter intimal injury show that leukocytes attach to the de-endothelialized surface of an arterial lumen within minutes.17,18,66 This includes polymorphonuclear neutrophil (PMN), monocytes, and lymphocytes. In addition to adhering to the damaged endothelium, these cells even penetrate it in some instances. These adherent leukocytes are paramount to the secretion of many cytokines and growth factors that influence the subsequent events in neointimal hyperplasia (Table 55-2).67–69 The central role of leukocyte chemotaxis in the arterial injury response has been demonstrated through several novel models of inflammation. One model that supports the role of inflammation in the development of neointimal hyperplasia consists of delivering leukocyte-derived myeloperoxidase in the presence of its substrate hydrogen peroxide (H2O2). The infusion of myeloperoxidase and H2O2 into an isolated rat common carotid artery without mechanical injury elicited the development of neointimal hyperplasia.70
TABLE 55-2 Cellular Sources of Growth Factors and Cytokines Involved in the Arterial Response to Injury
| Growth Factor/Cytokine | Leukocytes | Monocytes/Macrophages |
|---|---|---|
| IGF-1 | X | |
| PDGF | X | |
| TGF-α | X | |
| TGF-β | X | X |
| VEGF | X | X |
| EGF | X | |
| FGF | X | |
| TNF-α | X | X |
| IL-1β | X | X |
| IL-4 | X | |
| IL-6 | X | |
| IL-8 | X | X |
| IL-10 | X | |
| IL-18 | X | X |
| MCP | X | X |
| IGF-1 | X | |
| PDGF | X |
IGF, Insulin-like growth factor; PDGF, platelet-derived growth factor; TGF, transforming growth factor; VEGF, vascular endothelial growth factor; EGT, epidermal growth factor; FGF, fibroblast growth factor; TNF, tumor necrosis factor; IL, interleukin; MCP, macrophage chemoattractant protein.
Adapted from Mitra AK, Del Core MG, Agrawal DK: Cells, cytokines and cellular immunity in the pathogenesis of fibroproliferative vasculopathies. Can J Physiol Pharmacol 83:701–715, 2005.
In another model, an endotoxin-soaked thread was placed on half of a rat femoral artery to produce an inflammatory response.71 This technique consistently caused a significant leukocyte infiltration, which occurred only on the treated side of the vessel. Histologic examination performed 14 days later revealed nonuniform neointimal lesions in which proliferating VSMC were located exclusively on the side of the lumen adjacent to the treated half of the arterial wall. Clearly, these animal models suggested a strong association between inflammation and neointimal hyperplasia. The mechanisms controlling this relationship are a target for therapeutic intervention.
After activation and adhesion to the damaged vessel lumen, leukocytes migrate into the arterial wall. In one study, 42 days after a denuding injury, leukocytes were shown to penetrate the arterial media and were seen deep within the hyperplastic lesions.72 Several different mechanisms trigger this leukocyte migration. VSMC and macrophages elaborate potent chemotactic factors for leukocytes, which could sustain continued leukocyte recruitment.73 Supporting this notion, serum containing medium that was conditioned from VSMC stimulated leukocyte migration. Other chemotactic agents for leukocytes include PDGF and, in some reports, factors released by fibroblasts. VSMC removed from atherosclerotic plaques expressed ICAM-1, suggesting a possible connection between these medial cells and leukocyte recruitment and activation.
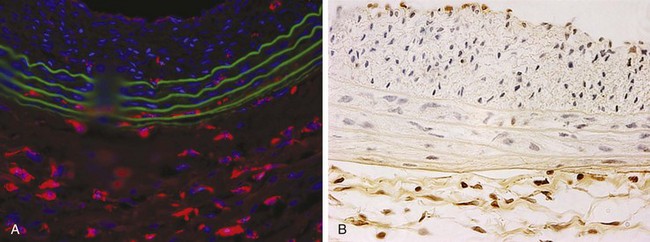
In addition to luminal entry, the adventitia has been recognized as an important site of inflammation after arterial injury (Figure 55-6). The accumulation of B cells, T cells, and macrophages in the adventitia has been documented in numerous studies.74–77 Okamoto and colleagues78 characterized macrophage localization to the adventitia after porcine coronary artery angioplasty and which receptors are important to their recruitment. Macrophages present in the adventitia were significantly increased at day 1 after injury, peaked at day 3, and then slowly decreased back to baseline at day 14. Interestingly, the number of adventitial macrophages was larger than the number found in either the media or the intima. In addition, macrophage accumulation preceded cell proliferation by 48 to 72 hours. Remarkably, they also found that macrophages had a nearly twentyfold greater presence than neutrophils. Early and sustained expression of E-selectin, P-selectin, and VCAM-1 by the vasa vasorum indicated that these receptors were important in allowing the macrophages to localize to the adventitia. Macrophages have also been noted to phenotypically differentiate into myofibroblasts after arterial injury and contribute to the development of neointimal hyperplasia.79 Bayes-Genis and coworkers79 demonstrated that 14 days after arterial injury to the pig coronary artery, 42% of the neointimal cells expressed the macrophage-specific antigen SWC3, and 9% of the cells co-expressed SWC3 and alpha smooth muscle actin (αSMA). Finally, macrophage localization and activation has been found to rely on angiotensin II (Ang II)–induced upregulation of CCL2 and IL-6. Tieu and associates was recently able to quantify the degree to which Ang II increases the macrophage population in the adventitia and found a 5.3-fold increase in populations of mice treated with Ang II.80 They also documented the importance that CCL2 plays in Ang II–induced macrophage localization because the absence of CCR2, a CCL2 receptor, in CCR2−/− mice significantly attenuated adventitial macrophage accumulation. The cytokine IL-6 was shown to be important to the activity of Ang II because IL-6−/− mice adventitia did not show the same propensity to recruit macrophages as the IL-6+/+ mice after Ang II stimulation.
Growth Factors
Angiotensin II, PDGF, and MDGF are all examples of growth factors believed to be involved in the formation of hyperplastic neointimal lesions; each has been reviewed in detail earlier. bFGF, a smooth muscle cell mitogen that is not secreted, is released from damaged VSMC and acts in an autocrine fashion to stimulate proliferation of adjacent, viable cells. This mitogen is largely responsible for the proliferative response after balloon injury.6 Denudation of endothelium with a fine wire that does not damage the endothelium results in much less VSMC activation.81 Lindner and associates19 observed that administration of bFGF after an injury increased VSMC proliferation from 11.5% to 54.8%. Their finding of an equivalent increase in proliferation after wire loop injury demonstrates that bFGF can act as a mitogen for undamaged medial cells. bFGF administered to normal, nondenuded arteries has no effect on VSMC growth. bFGF is also a potent stimulator of endothelial cells and, accordingly, stimulates angiogenesis. After arterial injury, bFGF is important for stimulating endothelial cell growth to completely cover injured regions that would normally remain denuded. Cuevas and colleagues demonstrated that the direct local infusion of bFGF into either normal adventitia or injured media results in proliferation of both vasa vasorum and VSMC.82
TGF-β is mostly known for its role in stimulating extracellular matrix deposition. More recently, however, it is now understood to have a role in negatively regulating endothelial cell activation, VSMC migration, and macrophage and lymphocyte accumulation.83 Early studies demonstrated that TGF-β was expressed after arterial injury.24 Furthermore, application of TGF-β to the arterial wall using gene therapy or intraluminal delivery stimulated the development of neointimal hyperplasia.23,84,85 Inhibition of TGF-β using antibodies inhibited neointimal hyperplasia.86–88 These early studies focused on the stimulation of procollagen and fibronectin, as well as effects on remodelling. However, identification of SMAD3 because the downstream target of TGF-β has shed more light on the precise role of TGF-β. SMAD3 knockout mice exhibited significantly more neointimal hyperplasia after injury compared with the wild type controls. Specifically, they exhibited more cellular proliferation, and bone marrow transplantation studies revealed that the neointima that developed in the SMAD3 knockout mice was derived from resident cells, not circulating cells.89
Renin-Angiotensin System
The concept that active Ang II in the vascular wall is synthesized locally, and not delivered via the systemic circulation, was originally suggested by Swales and Thurston.90 In this report, the authors noted that the amount of angiotensin antiserum required to inhibit endogenous angiotensin effects in sodium-loaded rats could not be explained solely on the basis of systemic production. In addition, several investigators had noticed residual renin-like activity after bilateral nephrectomy in animal studies.91 More convincing evidence for the existence of a locally active vascular renin-angiotensin system, operating independently of the classic systemic circuit, has been provided by several studies. Angiotensinogen mRNA, the only known precursor to the angiotensin peptides, has been detected in several extrahepatic vascular tissues, including the aorta.92,93 Immunohistochemical studies have demonstrated renin in cells throughout the vascular wall.94 Both VSMC and endothelial cells can synthesize renin in vitro.95,96 Also, mRNA coding for renin has been identified in human VSMC.91,97 Finally, ACE is located primarily on the luminal surface of vascular endothelial cells.98 Thus, the normally functioning vascular wall possesses all the necessary components for the independent local production of Ang II.
The physiologic function of locally produced Ang II is an area of continued controversy. Ang II receptors have been identified on vascular endothelial cells, VSMC, and circulating platelets.91,99–101 Ang II stimulation of endothelial cell–bound receptors leads to the secretion of prostacyclin and possibly endothelium-derived relaxant factor.102,103 Both of these substances cause VSMC quiescence and relaxation. Activation of the angiotensin receptors located directly on the VSMC themselves causes the opposite effect. Campbell-Boswell and Robertson104 reported that Ang II stimulates the proliferation of human VSMC in vitro. Geisterfer and associates105 found that the protein content of these VSMC increased by 20% when stimulated by Ang II for 4 days. Further, this stimulation was abolished by the Ang II receptor antagonist saralasin. Finally, the Mas protooncogene, located on the surface of medial VSMC, increased mitogenic activity when stimulated and has been identified as a functional Ang II receptor.106 Therefore, one explanation for the physiologic function of a locally active renin-angiotensin system is the autocrine balance of vascular wall metabolic activity and tone.
This view has led many investigators to study the effect of the local production of Ang II on the subsequent development of intimal hyperplasia. In this hypothesis, denudation of the arterial endothelium disrupts the balance of the local renin-angiotensin system and allows the anabolic and mitogenic effects of Ang II on the medial VSMC to proceed unchecked. Another possible mechanism for the promotion of hyperplastic neointimal growth by the local production of Ang II involves the activation of platelet metabolic pathways. As mentioned previously, human platelets possess specific binding sites for Ang II, and platelet activation and the subsequent release of PDGF may play a role in the development of neointimal hyperplasia.101 In a 1990 study by Swartz and Moore,107 Ang II enhanced both collagen-induced platelet aggregation and the production of thromboxane A2. Finally, the expression of PDGF by activated VSMC is upregulated in the presence of Ang II.108
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


