Pneumocystis Pneumonia
Despite widespread use of effective antimicrobial prophylaxis in susceptible immunocompromised patient populations, Pneumocystis jiroveci remains an important opportunistic pathogen. The incidence of Pneumocystis pneumonia (PCP) has decreased with the appropriate deployment of antimicrobial prophylaxis in susceptible hosts and with the advent of highly effective antiretroviral therapy (HAART) in HIV-infected individuals. PCP remains, however, an important syndrome in HIV infection in the developing world with high mortality. Pneumocystis also impacts the growing population of immunocompromised individuals following organ and hematopoietic stem cell and bone marrow transplantation, and with the broader use of immunosuppressive therapies in connective tissue, cancer, and immune disorders. The change in nomenclature this past decade, renaming of Pneumocystis carinii as P. jiroveci, reflects knowledge about the organisms responsible for this syndrome in different host species. This change has generated significant controversy among scientists, clinicians, and journal editors. Advocates and detractors alike agree that no matter the species name, Pneumocystis pneumonia or PCP should continue to be used to describe disease caused by this organism in humans.
HISTORY AND BACKGROUND
The cyst form of Pneumocystis was first described in 1909 by the famous parasitologist, Chagas and in 1910 by his colleague Carinii, and was described as a parasite and part of the life cycle of Trypanosoma cruzi.1 The organism was first associated with pulmonary infection in rats by Delanoe and Delanoe but was not recognized in humans until 1942 (Van der Meer and Brug) and was not associated with human disease until 1952 (Vanek and Jírovec) when it was found in association with “plasma cell interstitial pneumonitis” among malnourished children and neonates.2 Small epidemics of plasma cell interstitial pneumonitis had been noted in children in orphanages in Europe in the 1930s and following World War II and the Vietnam War. In the 1950s, recognition of congenital immune deficiencies and the development of immunosuppressive therapies identified new hosts for PCP. At that time, PCP was recognized in patients receiving corticosteroids and chemotherapeutic drugs and in immunosuppressed rats receiving corticosteroids.
The increasing incidence of PCP led to epidemiologic and therapeutic studies of the disease by the Centers for Disease Control (CDC) in the 1970s, based on the provision of the sole therapeutic agent available at that time (pentamidine). Clusters of P. carinii were reported at a variety of clinical oncology and transplant centers. However, the development of pyrimethamine, sulfadoxine, and of trimethoprim (TMP) and sulfamethoxazole (SMX) for the treatment and prevention of Pneumocystis infection greatly reduced the occurrence and the morbidity of the infection. These agents are now generally used in a fixed combination (TMP-SMX, or cotrimoxazole). P. carinii became known the world over as being the first disease-defining illness associated with AIDS in the 1980s, causing over one-fourth of community-acquired pneumonias in HIV-infected persons and more than 200,000 cases of PCP since 1979. What was once an obscure, poorly understood opportunistic disease, became a common clinical entity in a matter of a decade as a result. By the mid-1990s genetic diversity among isolates of Pneumocystis from different host species had been described suggested that the organism was a fungus rather than a parasite and that there was host species specificity. The basis of that specificity (i.e., receptors, nutritional requirements) remains unknown. The organism causing infection in humans was renamed P. jiroveci after the Czech parasitologist, Otto Jírovec.3
STRUCTURE AND LIFE CYCLE
In humans and animals, three forms of the organism have been identified: trophozoite, cyst, and sporozoite (or intracystic bodies) (Fig. 135-1). The trophozoite, 2 to 5 μ in diameter, is either round or sickle-shaped and contains a nucleus, mitochondria, and vacuoles. It also includes pseudopodia and filopodia, used in limited motility. The cyst usually measures between 3 and 6 μ in diameter. Its cell wall consists of three layers, and its cytoplasm contains eight small pleomorphic intracystic oval bodies called sporozoites.4 Two other cystic forms have been described, but these are probably intermediates including empty or developing cysts (Fig. 135-1). Many small surface projections, or tubular expansions, form a branching network over the surfaces of the cysts and the trophozoites.
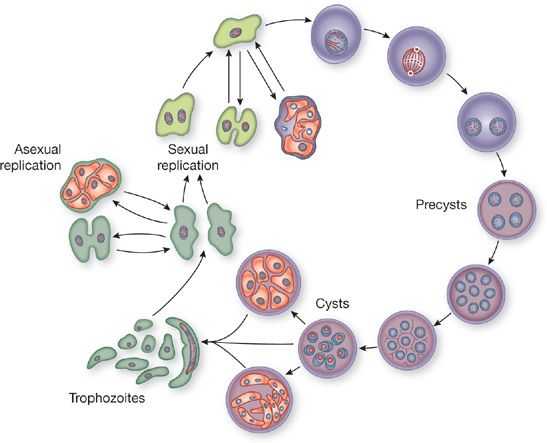
Figure 135-1 The life cycle of Pneumocystis carinii.
In the alveolus, Pneumocystis are covered with a variety of glycoproteins derived from both the organism and the host. Specific and nonspecific immunoglobulins, albumin, surfactant proteins, laminin, fibronectin, and other serum and lung proteins coat the surface. The organism produces a relatively limited array of surface glycoproteins that share antigenic epitopes. These, and some minor components, are found on both animal- and human-derived organisms. Pneumocystis from different species share antigenic epitopes in addition to carrying unique epitopes.5 The cell wall contains cholesterol but no ergosterol and does not appear to synthesize sterols de novo; this likely accounts for the lack of susceptibility to the azole and polyene antifungal antibiotics.6 The presence of chitin in the cell wall is controversial. The surface of P. jiroveci is carbohydrate-rich with glucose, mannose, and β-1,3-glucan, the latter of which may play a role in phagocytosis of the organism by macrophages and may provide a potential diagnostic and therapeutic target for echinocandin antifungal agents. The surface also contains carbohydrate-binding moieties, which may play a role in attachment to epithelial or surfactant layers.
The life cycle of Pneumocystis is incompletely understood (Fig. 135-1). Many of the studies of the life cycle have used organisms derived from infected animals and passaged on a feeder cell layer of epithelial or fibroblastoid cells in tissue culture. Continuous growth has not been achieved in this system, and the human-derived organisms have not been grown consistently in vitro. Success with axenic or cell-free cultivation of P. jiroveci has been limited. It is now believed that the sporozoites, or daughter forms, emerge from the cyst to develop into trophozoites. These trophozoites mature to form cysts and then repeat the cycle.7 This sequence is far from settled, however, and both sexual and asexual intermediate stages have been postulated. It is likely that some differences exist in Pneumocystis growth in different hosts and with different immune defects.
Research on Pneumocystis has been hampered by the difficulties encountered in propagating the organism in vitro. Studies have been performed on organisms derived from immunosuppressed rodents, which develop PCP in the absence of prophylaxis or respiratory isolation. Researchers have used this model to demonstrate the aerosol transmission of P. jiroveci.8 To this day, unfortunately, cell culture techniques remain useless diagnostically because of the difficulty in culturing the organism from the infected human tissues.
TAXONOMY AND MOLECULAR BIOLOGY
Phylogenetic data support the identification of P. jiroveci with the taxonomic kingdom of fungi (Rhizopoda, Myxomycetes, Zygomycota, Schizosaccharomyces, Neurospora, Candida, and the red yeasts in various studies), rather than the parasitic kingdom of protozoa. This is based on conserved mRNA sequences and other findings.9 The presence of separate genes encoding the thymidylate synthase and dihydrofolate reductase of P. jiroveci, the presence of a cyst wall rich in β-glucan that stains with periodic acid–Schiff and silver stains, the poorly developed mitochondria, the absence of typical protozoan intracellular organelles, and the airborne spread of infection all support this taxonomic position. The neutral lipid fraction of P. jiroveci includes a variety of phytosterols shared by plants and fungi, including Physarum species.10 However, the appearance of the organism with a thick-walled cyst with internal sporozoites and ameboid trophozoites, the absence of ergosterol, susceptibility to antibiotics used in the treatment of protozoan infections (pentamidine, atovaquone, SMX), and the existence of antigenic variation in the major surface glycoprotein (gp120, gpA, MSG) lend credence to the original identification with the protozoa.
Complicating the taxonomy of this organism is the existence of different strains of Pneumocystis that have been demonstrated using pulsed-field gel electrophoresis and DNA sequencing.11 This diversity is greatest between strains infecting different host species, giving rise to the eventual renaming of the organism.12 It appears that many infections in individuals are often clonal, though multiple strains may coexist in any one single infected person (see below).11
P. jiroveci expresses both unique and some common antigens in different host species. Surface antigens have been characterized at the glycoprotein and molecular levels. The MSG represents the main humoral immunogen in the rat model, although other antigens (gp45–55) may have importance in human infection.13 Several MSG types have been observed simultaneously in single infected humans and animals with monoclonal antibody staining and genetic characterization. The MSG appears to represent a large family of related genes (approximately 50–80), many of which are located in tandem repeated arrays in the subtelomeric regions and may contribute to the generation of the variety of antigenic types. Cross-reactive T cell responses to MSG variants is uncommon and suggests the possibility that Pneumocystis utilizes MSG switching to evade human immune responses.14
EPIDEMIOLOGY OF INFECTION DUE TO PNEUMOCYSTIS
Over the past 60 plus years, PCP has been transformed from a medical curiosity into an important respiratory infection that affects four categories of immunocompromised host: (1) congenital, caused by inborn immune defects in antibody-synthesizing capacity and/or the cellular mechanisms responsible for delayed hypersensitivity; (2) induced, by immunosuppressive therapy, especially corticosteroids (3) acquired, occurring as an identifying opportunistic pathogen in HIV infection or with exogenous immune suppression; and (4) nutritionally deficient with epidemic infection, described primarily in susceptible neonates and infants.
P. jiroveci causes pneumonia in persons with a wide variety of underlying immune deficiencies (Table 135-1). Studies performed in immunosuppressed animals and clinical experience indicate that T cell immune defects predominate in individuals with P. jiroveci infection. In HIV infection, the progressive depletion of CD4+ T cells heralds the onset of risk for PCP, though CD8+ T cells likely play an important role in responding to Pneumocystis infection. Passive transfer of immune CD8+ effector T lymphocytes is protective against PCP in mice.15 Other immune defects such as neutropenia and hypogammaglobulinemia enhance the risk of PCP by contributing to the overall net state of immunosuppression, and disease may be seen in patients with persistent defects in leukocytes and antibodies, but appears less important for protection than is cellular immunity. Passive immunoglobulin transfer in mice does not have a significant protective effect.16
TABLE 135-1 Conditions Associated with Pneumocystis Pneumonia
The relative risk of infection with Pneumocystis is predictable in most hosts in which infection occurs, including the non-transplant and non-HIV infected populations (Table 135-2). The risk of PCP is greatest in the first 6 months after solid-organ transplantation, after 3 to 6 months of oral corticosteroid therapy (in excess of 15–20 mg prednisone equivalent), and during periods of intensified immune suppression, as with the use of bolus corticosteroids or lymphocyte-depleting antibody therapies for graft rejection.17 Disease occurring outside this host population or predicted timeframe should suggest an excess epidemiologic hazard or unidentified immune deficiency. PCP has also complicated the syndrome of rapamycin lung, an idiosyncratic syndrome of diffuse pulmonary infiltrates in solid-organ transplant recipients receiving sirolimus-based immune suppression. In untreated AIDS, the risk for PCP increases with the progressive fall of the CD4+ lymphocyte counts to below 200 cells/mm3 or to less than 20% of the total lymphocyte pool. The correlation with T-lymphocyte numbers is such that the rate of infection nearly doubles with a drop in CD4+ lymphocyte counts from between 100 and 200 to below 100/mm3.18 The presence of thrush has also been shown to be an independent predictor of risk for PCP in patients with HIV as has a previous bout of PCP. Rarely, PCP occurs at CD4 counts greater than 200 cells/mm3 and prophylaxis should be offered to such patients starting around the level at which PCP occurred. The occurrence of PCP infection in persons not in these categories should suggest exposure to infected persons, other immunosuppressive effects (e.g., coinfection with cytomegalovirus (CMV), lymphoma, neutropenia), or in the HIV-infected person, a rapid progression of viral infection with the accompanying decline in systemic immune function.
TABLE 135-2 Reported Attack Rates for PCP in Non-Transplant Recipients and Non-HIV-Infected Patients Not Receiving Prophylaxis by Underlying Condition
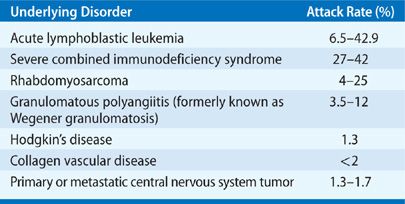
The spectrum of patients developing PCP has changed with the advent of HAART for HIV infection and the routine use of prophylaxis for most patients with hematologic malignancies and after organ transplantation. Inconsistent use of prophylaxis, however, has been linked to some of the published outbreaks of PCP in solid-organ transplant recipients in recent years.19 In most HIV-negative, immunocompromised patients, the risk of disease is of the order of 5% to 15%, though the risk following organ transplantation varies with the immunosuppressive regimen and the organ transplanted. The attack rate is highest in the lung and combined lung–heart recipients.20 The major risk factors for PCP are reflected in a retrospective study of 116 HIV-negative patients with PCP of which 30.2% had hematologic malignancies, 25% were organ transplant recipients, 22.4% had inflammatory disorders, 12.9% had solid tumors, and 9.5% had other conditions. Corticosteroids use was reported in 90.5% of these patients. The median daily dose was 30 mg of prednisone; however, 25% of the patients had received as little as 16 mg/day of prednisone. The median duration of corticosteroid therapy before the diagnosis of PCP was 12 weeks. However, 25% of the patients developed PCP after 8 weeks or less of corticosteroid use.21 Ultimately, specific immunosuppressive drugs may be less important than the overall net state of immunosuppression that can be engendered by multiple factors (metabolic factors, age, immune cell quantity and quality, viral coinfection, underlying lung disease).
In children, epidemiologic studies suggest that PCP may occur in the setting of concomitant viral infection and may be found in infants who die unexpectedly in the community. Beard et al. demonstrated that different strains of Pneumocystis appeared to be present in specimens obtained from infants compared with those from HIV-infected adults. This suggests that different strains circulate among non–HIV-infected infants and in HIV-infected adults.22 The basis for this difference remains unclear and requires confirmation among larger populations.
The natural reservoir of infection remains unknown, though Pneumocystis species are thought to be fairly ubiquitous in nature. Aerosol transmission of infection has been demonstrated in animal models, and clusters of infection have developed in clinical settings. Outbreaks among solid-organ transplant recipients and among HIV patients have been documented in hospital wards.19,23–27 Molecular typing studies have implied that some of these outbreaks could be due to person-to-person transmission.19,23,24 P. jiroveci DNA has also been detected by polymerase chain reaction (PCR) in the air of hospital rooms, bronchoscopy suites, and clinics used by infected subjects.28,29 Whether or not these outbreaks are due to person-to-person transmission or some common environmental source remains unclear. These data have led some experts to recommend strict hospital segregation of immunocompromised hosts with PCP and the use of facemask filtering in the clinical setting, but the efficacy or need for these approaches has not been demonstrated. Prophylaxis remains likely the most effective method at preventing infection in any setting where patients may be at risk of disease.
The prevalence of infection with P. jiroveci in patients with AIDS prompted a large effort to study the epidemiology of PCP more closely by developing diagnostic serologic tests that might be applied to the general population. Serologic testing reinforced the view that subclinical infection, exposure, or colonization is common. Most people have serologic evidence of exposure by age 4 years.30 Other studies have uncovered the presence of Pneumocystis in the respiratory tract of asymptomatic nonimmunocompromised hosts. One such study found that as many as one in five people may have Pneumocystis DNA in bronchoalveolar lavage (BAL) fluid.31 These data, along with animal models, have supported the possibility of reactivation of previously established infection as being a potential cause of disease in susceptible hosts. However, data regarding outbreaks, evidence that the infection can be eliminated, subsequent infection with distinct genotypes, and the geographic variability in disease, all combine to suggest that alternative modes of transmission besides reactivation of latent disease is predominant.
In HIV patients, a clear correlation with circulating HIV viral loads is not well established. Historically, the rate of PCP in AIDS was halved by the use of zidovudine for the duration of the effective antiviral effect of this agent.32 More pronounced effects have been observed in individuals who regain immune function for more than 6 months under HAART therapies; such patients may be able to avoid primary prophylaxis. The rate of infection appears to double in men who have sex with men with AIDS when compared with intravenous drug users. In the absence of effective prophylaxis or HAART, more than 80% of AIDS patients are expected to develop PCP.18 Clinically symptomatic infection may emerge during the weaning of immunosuppressive agents or as a part of the immune reconstitution syndrome seen with HAART in AIDS. This is consistent with the limited inflammatory response generated by the organism in the susceptible host.
CLINICAL PRESENTATION
The hallmark of infection due to P. jiroveci is the presence of marked hypoxemia, dyspnea, and cough out of proportion to physical or radiologic findings. In the transplant recipient or the person undergoing corticosteroid therapy, PCP is generally acute-to-subacute in development and often masked by other processes, including allograft rejection, graft dysfunction or secondary infection. In the AIDS patient with a first episode of infection, the evolution is more gradual (often 2–5 weeks) and constitutional symptoms such as weight loss can be prominent. Subsequent infections may evolve more rapidly. In the cancer patient receiving chemotherapy or in the organ or bone marrow–stem cell transplant recipient, the use of corticosteroids, poor lung infection, abnormal pulmonary lymphatics (after heart, lung, or liver transplantation), and neutropenia may contribute to the absence of radiologically apparent disease. The rate of development of clinical infection is exacerbated by the presence of pre-existing lung disease (e.g., cyclophosphamide lung or pulmonary fibrosis) or other infections (e.g., CMV, Legionella, mycobacteria) (Fig. 135-2). In the organ transplant recipient, PCP most frequently occurs approximately 2 to 4 months after the initiation of immune suppression or during periods of increased immune suppression (pulsed corticosteroids, antilymphocyte therapies, CMV infection). The incidence and timing of infection vary between institutions, however, and can be dependent on the prophylactic regimens employed. In some series of patients receiving lung or heart–lung transplants the rate of asymptomatic isolation of P. jiroveci can be higher than expected, approaching 10% or higher.33,34
In AIDS patients without HAART, the presentation of PCP is often complicated by a variety of factors. Prophylaxis with aerosolized pentamidine or subtherapeutic levels of other antibiotics (e.g., dapsone, atovaquone) may delay or alter the presentation of disease and confound diagnostic assays. As in most immunocompromised hosts, coinfection with other organisms may accelerate the progression of disease or alter the radiographic pattern; in particular, CMV, Histoplasma capsulatum, Legionella spp., and mycobacterial species may contribute to the constitutional symptoms, hypoxemia, and the localization of pulmonary lesions.
Acute exposure to Pneumocystis is rarely documented. In animal models, inoculation of P. jiroveci induces a neutrophilic infiltrate that is rapidly (2–3 days) replaced with lymphocytes and macrophages. Similarly, in transplant recipients and despite therapy with cyclosporine, Pneumocystis induces lymphocyte- and neutrophil-predominant infiltration into the lungs acutely, followed by macrophage infiltration and clearance of organisms. This contrasts with the macrophage- and neutrophil-predominant infiltrates seen in AIDS patients with acute disease. The lymphocytes are primarily T lymphocytes with normal CD4/8 ratios. Coinfection with CMV and other pathogens will be detected in more than half of Pneumocystis-infected patients and likely play a role in the individual immune response of each patient.21,22 The nature of the immune suppression determines the types of other opportunistic pathogens that coinfect the compromised host and the overall risk of PCP. When cyclosporine began being used widely as an alternative immunosuppressant agent to azathioprine in solid-organ transplantation, the incidence of PCP increased among susceptible transplant recipients.35 The subsequent development and use of tacrolimus seemed to increase the risk further.36 The net state of immunosuppression these complex patients experience appears to play a bigger role than any single agent.
EXTRAPULMONARY PNEUMOCYSTOSIS
Pneumocystis was known to cause extrapulmonary disease in the pre-AIDS era, but metastatic infections have been most commonly observed in patients with untreated AIDS. Even in the untreated AIDS population, the incidence of extrapulmonary Pneumocystis infection is probably less than 1% of cases.37 It has been thought that susceptible AIDS patients might be more likely to develop extrapulmonary disease while on inhaled pentamidine prophylaxis since this modality of prevention is largely lung-specific and does not achieve significant systemic drug levels like TMP-SMX. The evidence for this is minimal and difficult to assess with a low incidence of extrapulmonary disease. The diagnosis of extrapulmonary disease is challenging, and clinicians must have a high suspicion to identify infection outside the lungs. Respiratory disease may be minimal or absent in patients with extrapulmonary infection. In addition to the liver and spleen, sites of extrapulmonary disease have included eye, ear, lymph nodes, thymus, skin, mastoids, ascites, gastrointestinal tract and omentum, pleura, kidney, bone marrow, pancreas, and adrenal glands, and it has been reported as a cause of thromboembolic disease. Vasculitis has been reported due to P. jiroveci as a cause of ischemic necrosis of the digits and has been associated with granulomatous inflammation on tissue biopsies.38
The presentation of extrapulmonary infection is generally a mass lesion with accompanying fever, sweats, and malaise. Visual loss may accompany retinal lesions, hepatitis with liver impairment, and ascites and gastrointestinal tract obstruction with peritoneal and omental lesions. By computed tomographic (CT) scan, many nonenhancing, low-attenuation masses, often with necrosis and/or hemorrhage, may be seen in the liver or spleen (Fig. 135-2). Calcification may occur at the edge of such necrotic lesions during the acute infection, often in the hepatic or renal parenchyma. Histopathology will demonstrate granulomas with giant cells, calcification, or cavities. Distant sites may also contain the same frothy hyaline material seen in the alveoli in pulmonary disease (Fig. 135-3). P. jiroveci may be seen adherent to the blood vessel walls with myointimal inflammation and thrombosis. Dual infections at extrapulmonary sites may occur with other opportunistic organisms just like in the lung, including mycobacteria, Histoplasma, Legionella, other fungi, or common bacteria. Unless superinfection has occurred or splenic rupture or other life-threatening condition is imminent, systemic treatment of multiple small abscesses due to Pneumocystis infection should be adequate without surgical intervention.
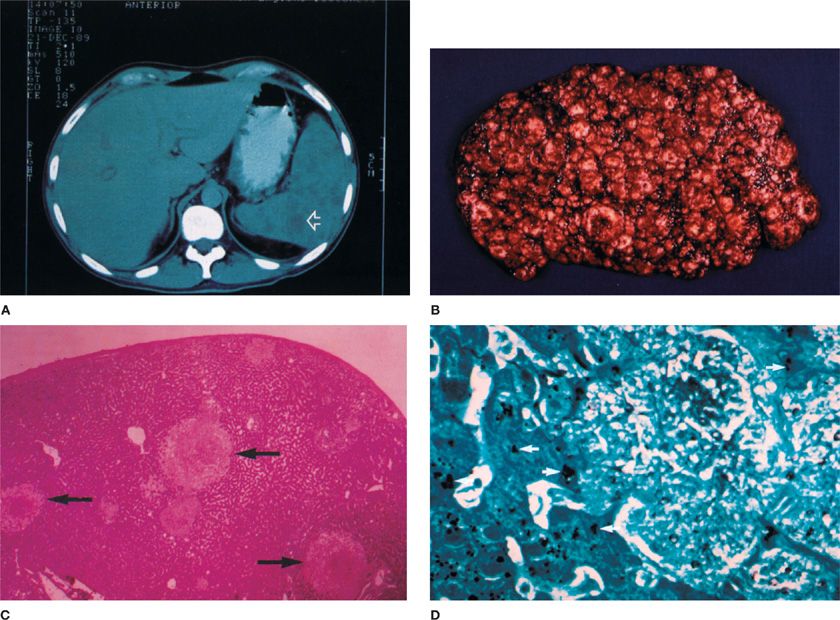
Figure 135-2 Extrapulmonary pneumocystosis in a patient with AIDS on aerosolized pentamidine prophylaxis presented as a splenic mass lesion with abdominal pain. The computerized body tomographic (CBT) scan reveals a mass lesion (A), seen after splenectomy on gross pathology (B,C) and on microscopic examination (silver stain, D). (Used with permission of Dr. J. Davis Allen, Jr., New England Deaconess Hospital.)
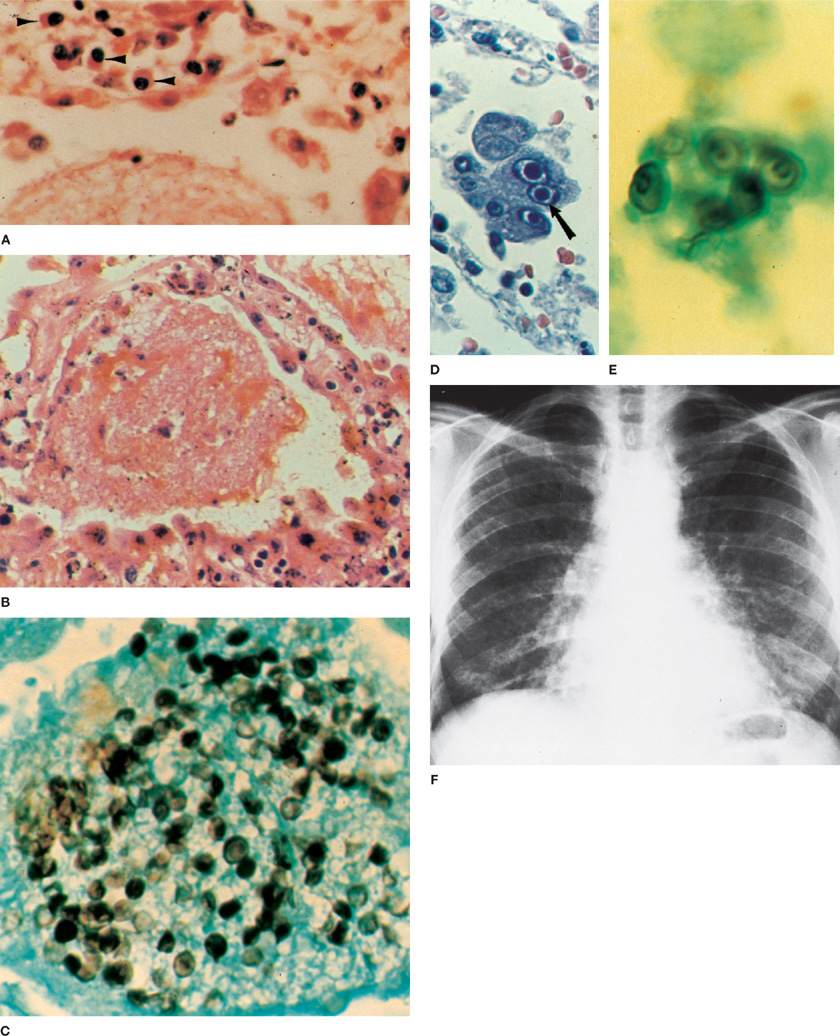
Figure 135-3 Pneumocystis jiroveci pneumonia. A. Lung of a malnourished infant showing an intra-alveolar foamy exudate and plasma cells (arrowheads) in the interstitium (H&E stain, × 620). B. Typical alveolar exudative pattern from the lung of an adult with Pneumocystis pneumonia after therapy with corticosteroids. Swelling of the alveolar epithelial cells and interstitial edema are seen. Inflammatory response in interstitium is minimal (H&E stain, × 500). C. Pneumocystis in the form of thick-walled cysts within the foamy exudate (displayed by Gomori’s methenamine-silver nitrate stain and brilliant green counterstain, × 1250). D. Cytomegalovirus inclusion bodies in alveolar macrophages in a patient with Pneumocystis pneumonia (H&E stain, × 720). E. Pneumocystis cysts in cytologic preparation of induced sputum from an AIDS patient (silver stain, × 1250). F. Pneumocystis pneumonitis. Typical chest radiograph showing bilateral, diffuse interstitial infiltrates extending from hilar area.
THORACIC IMAGING
Imaging modalities useful in clinical assessment for PCP include chest radiography, nuclear medicine techniques, and CT scanning.
 THE CHEST RADIOGRAPH
THE CHEST RADIOGRAPH
The chest radiograph plays a central role in the diagnosis of PCP (Figs. 135-4 and 135-5). No radiographic pattern is pathognomonic for Pneumocystis infection. PCP presents with hypoxemia and fever without significant sputum production and is more often than not, a diffuse interstitial process on chest radiograph.39 The radiographic pattern ultimately depends on the patient’s underlying or accompanying disease, state of immunosuppression, and duration of infection. Sometimes the chest radiograph is normal despite overt pulmonary disease. More often, the early stage of PCP is manifested by fine, bilateral, perihilar, diffuse infiltrates that progress to an interstitial alveolar butterfly pattern; from the hilar region, the infiltrates often spread to the apices or bases. Despite therapy, this pattern can often be succeeded in 3 to 5 days by progressive consolidation, the appearance of air bronchograms, and complete opacification of the lung fields. Unusual courses and patterns are often observed: nodules, unilateral infiltrates, or even lobar consolidations right from the start (Figs. 135-4 and 135-5). Small pleural effusions may also occur. Distortions in pattern are commonly produced by prior irradiation, drug-induced pulmonary injury, or concurrent infections with other organisms. The patient with recurrent disease may develop chronic interstitial markings, small cysts, or honeycombing on chest radiograph. The distribution of cysts in pneumocystosis, when present at all, is more often diffuse, whereas peripheral or apical bullae are often seen without infection in intravenous drug abusers. Rarely, cavitary disease is seen in the absence of other pathogens; however, P. jiroveci can superinfect fungal or mycobacterial cavities.40
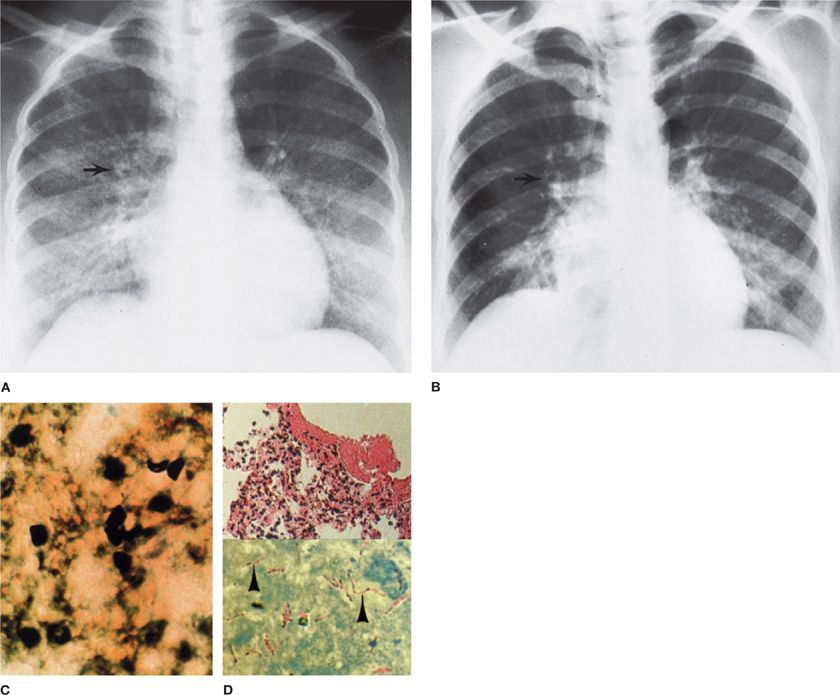
Figure 135-4 Atypical pattern of Pneumocystis with Mycobacterium avium complex in a Haitian woman with AIDS. A. Diffuse pulmonary infiltrates before the treatment of Pneumocystis infection. Arrow indicates small abscess cavity. B. After the treatment for Pneumocystis, many small cavities persist (arrow). C. Transbronchial lung biopsy of initial infiltrate reveals Pneumocystis cysts (silver stain, ×760). D. Open lung biopsy after therapy for P. jiroveci included areas of pneumonitis atypical for Pneumocystis (upper), which contain M. avium-intracellulare (arrowheads, lower) (Kinyoun acid-fast stain, × 950).
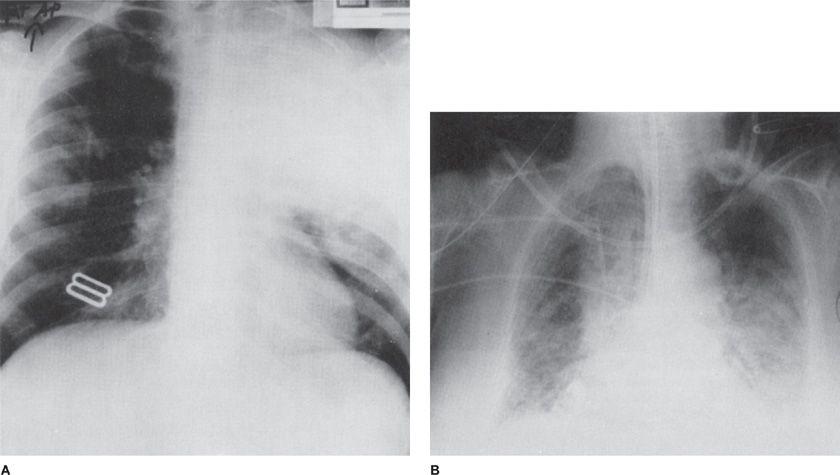
Figure 135-5 Atypical pneumocystosis. A. Upper lobe pneumonia in an AIDS patient on prophylactic aerosolized pentamidine therapy for a history of Pneumocystis pneumonia. The patient had both P. jiroveci and Legionella pneumophila infections. B. Persistent pulmonary infiltrates in a patient undergoing chemotherapy for non-Hodgkin lymphoma. Despite 21 days of therapy for P. jiroveci, oxygenation and pulmonary infiltrates failed to improve. An open lung biopsy revealed cytomegalovirus pneumonitis (which responded to therapy with ganciclovir) and drug-induced interstitial fibrosis.
The use of aerosolized pentamidine for the therapy and prophylaxis of PCP in AIDS created some new problems in the diagnosis and treatment of pneumocystosis (Fig. 135-5). Breakthrough disease in patients receiving aerosolized pentamidine classically presents largely or solely in the upper lobes on chest radiograph. Similar presentations may be seen without pentamidine use, suggesting a predilection of infection for the upper lobes. Pneumothorax is another classic finding associated with PCP. Evaluating this particular complication is challenging, but in AIDS patients may be seen in close to 5%. Most pneumothoraces are spontaneous in nature, but roughly half can also be attributed to mechanical ventilation and complications of other procedures. Underlying cystic disease may be a contributing factor to this phenomenon and it may cause significant morbidity and mortality, particularly in patients already requiring mechanical ventilation.41
In lung transplant patients, both rejection of the lung allograft and infection often produce similar abnormal chest radiographs. In the first month after transplant, rejection of the allograft lung will cause radiographic changes in up to 75% of patients. These changes include nodular and interstitial infiltrates in the perihilar area and the lower lobes, which may progress to consolidation. These changes may also occur with infection, of which CMV is not uncommon. CMV may be indistinguishable from PCP or organ rejection without biopsy. After the first month, rejection less often yields radiographic changes (about 25%), and the radiographic findings of infection are more often similar to those of other immunocompromised hosts.
 NUCLEAR IMAGING
NUCLEAR IMAGING
Nuclear medicine scans are generally nonspecific and add little to the diagnosis of pulmonary pneumocystosis. However, a normal scan will generally exclude diffuse pulmonary infection due to Pneumocystis. Gallium citrate, technetium, indium-immunoglobulin, white blood cell, and diethylenetriamine penta-acetate (DTPA) scans are abnormal in more than 90% of patients with Pneumocystis infection. These may be most useful in following the resolution of infection. Gallium scintigraphy can be entirely normal for patients with opportunistic pneumonia with or without AIDS. Conversely, AIDS patients may have abnormal gallium scans in the absence of other infection. Gallium scans often become abnormal before the radiographic appearance of pulmonary disease. Lymph node uptake alone may indicate the condition formerly referred to as AIDS-related complex (ARC), due to HIV infection. The pattern of lymphoid interstitial pneumonitis (LIP) in children with AIDS is indistinguishable from that of PCP as well.42 Diffuse pulmonary uptake may indicate occult infection in the asymptomatic patient, which would allow early intervention for therapy or prophylaxis, but the cost of routine gallium scanning for AIDS patients outweighs utility when compared with the judicious use of antiviral or anti-Pneumocystis antibiotics based on CD4 cell counts, viral load measurements, or other clinical data, and are generally not used for these reasons.
The role of other nuclear medicine–based imaging scans remains unclear. 18F-fluorodeoxyglucose (FDG) positron emission tomography (PET) scan imaging has been evaluated in a few small series. Abnormal uptake in the lungs has been seen in confirmed cases of PCP and has even been documented prior to the development of other abnormalities on routine chest radiography.43 Thus, PET scans may be useful as an early diagnostic tool, although lacking the specificity required for the diagnosis and guidance of specific therapy.
 COMPUTED TOMOGRAPHY AND OTHER RADIOGRAPHIC DIAGNOSTIC TECHNIQUES
COMPUTED TOMOGRAPHY AND OTHER RADIOGRAPHIC DIAGNOSTIC TECHNIQUES
Sometimes, early in the disease process, chest CT may demonstrate abnormalities not appreciated on routine chest radiography. Among HIV patients, the most frequent pattern early in the disease process will be a ground-glass opacification in the central/hilar and apical regions of the lungs bilaterally.44 In non–HIV-infected immunosuppressed patients, the opacification patterns may be more dense and distorting of the underlying lung architecture.45 Resolution of abnormalities can be quickly appreciated by CT imaging as well in patients receiving appropriate therapy. Other imaging modalities, such as magnetic resonance imaging (MRI) scans and ultrasound imaging are probably better suited defining extrapulmonary lesions such as pneumocystomas occurring in the soft tissues or intra-abdominal organ systems (discussed above; see Fig. 135-2). Their role in isolated pulmonary disease remains undefined.
LABORATORY FINDINGS
A number of nonspecific indicators of pulmonary processes have been used in the presumptive diagnosis of PCP. In general, the patient will have a PO2 less than 60 mm Hg and a respiratory alkalosis. The serum lactic dehydrogenase (LDH) enzyme will be elevated in almost all cases of PCP (over 300 IU/mL). Because LDH is likely a marker of underlying lung injury in PCP, the degree of elevation correlates with underlying hypoxemia and may have prognostic value.46 Unfortunately, LDH is nonspecific, and should generally be avoided as a diagnostic tool alone in cases of PCP. Lymphoma, hemolysis, other infections, ischemia, and LIP may also raise the LDH level. Respiratory distress and respiratory failure requiring intubation carry a poor prognosis. The marked hypoxemia of PCP is accompanied by a PAo2–PaO2 gradient rise; gradients over 30 mm Hg at the start of therapy are associated with a high mortality (and are an indication for the use of adjunctive corticosteroid therapy—see below). Both LDH and the arterial oxygenation gradient will return to normal with successful therapy. Another nonspecific indicator of lung injury in PCP is the angiotensin-converting enzyme (ACE) level (also nonspecific in that it can be elevated by smoking and sarcoidosis, among other causes). KL-6, a mucin-like glycoprotein expressed on pneumocytes and bronchiolar epithelial cells can also be detected in the serum of patients with PCP, but once again is likely a nonspecific marker of pneumonitis in general.46 While pulmonary function testing may reveal abnormalities in oxygen exchange and compliance in PCP, they are not useful diagnostically in the acute setting.
Recent interest has been garnered by the plasma-based (1,3) β-D-glucan assay. (1,3) β-D-glucan is a polysaccharide monomer found in a number of fungal cell walls including Pneumocystis species. It initially gained use as diagnostic assay in other forms of invasive fungal infections including candidiasis, aspergillosis, and fusariosis.47 Other opportunistic fungi, like Mucor species and Cryptococcus neoformans have little-to-no (1,3) β-D-glucan in the cell walls and the assay has not been useful for these infections. The presence of (1,3) β-D-glucan in Pneumocystis cell walls, however, makes this assay an attractive noninvasive laboratory test for PCP. A number of case series have examined the use of the assay and found a high sensitivity (>90%).48–54 Because β-D-glucan can be found in other fungal infections, the specificity is lower (less than 80%) in an unselected population. False positive assays occur with immunoglobulin and albumin therapies, and after hemodialysis and exposure to gauze. The assay also has limitations as a marker of response to therapy. Small retrospective evaluations have shown that some patients actually experience serum elevations of (1,3) β-D-glucan while on therapy and showing clinical improvement.55 The utility of this assay in determining prognosis or in therapeutic monitoring is unclear.
Given the ubiquity of the organism in the environment and the finding that the majority of children have detectable antibodies, use of serologic testing in the diagnosis of PCP is not recommended. Serologic testing may be complicated by inadequate antibody production in many immunosuppressed patients producing a lack of specificity and sensitivity in the diagnosis of acute disease.
SPUTUM EXAMINATION AND HISTOLOGIC DIAGNOSIS
Despite advances in imaging and laboratory testing, the gold standard for the diagnosis of PCP remains the identification of characteristic organisms on examination of pulmonary specimens (Table 135-3). The organism cannot be routinely cultured in the clinical laboratory, and thus direct visualization and correct identification are required. The methods used in making the diagnosis have been changed by the use of the induced sputum examination and bronchoalveolar lavage (BAL) without biopsy, and by immunofluorescent staining using monoclonal antibodies to P. jiroveci. As a result, the morbidity associated with diagnostic procedures has been reduced at the same time the diagnosis of coinfecting pathogens has been reduced.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


