Platelet Inhibition, Anticoagulants, and Thrombolytic Therapy
Anthony J. Comerota
Teresa Carman
Vascular surgeons have pushed the limits of technical expertise in terms of revascularizing the ischemic lower extremity. However, technical success can be undermined by hypercoagulable states, neointimal fibroplasia, high-resistance outflow beds, and progressive disease. Thrombotic complications, both primary and secondary, are conditions common to the practice of patients with vascular disease. Pharmacotherapeutic manipulation has become increasingly important in the ongoing management of patients with vascular disease, on both the arterial and venous sides of the circulation.
Platelet inhibition is a mainstay of the treatment of all patients with atherosclerotic disease. The use of lower-dose aspirin and “second-generation” platelet inhibition has demonstrated significantly improved results compared to traditional therapy.
Anticoagulation is constantly being refined for the management of patients with venous thromboembolic disease. Patients treated for idiopathic venous thromboembolism have consistently benefited from longer duration of anticoagulation.
A scientifically engineered pentasaccharide has demonstrated significant improvement in the prevention of deep vein thrombosis (DVT) in high-risk orthopedic patients. It is being studied for the treatment of established thrombotic disorder and in patients with atrial fibrillation.
Direct thrombin inhibitors are now the treatment of choice for patients with heparin-induced thrombocytopenia and may offer attractive treatment alternatives to patients with established thrombotic disorders.
Once thrombosis occurs, a strategy to remove the thrombus with catheter-directed thrombolysis for major arterial and venous thrombosis and systemic thrombolysis for pulmonary embolism has offered substantial potential benefit to our patients. Eliminating the underlying thrombus, restoring cardiopulmonary hemodynamics, and identifying and correcting an underlying arterial or venous stenosis offer long-term benefit and improved quality of life.
This chapter discusses the commonly used pharmacotherapeutic agents for the management of patients with vascular disease and presents a brief overview of their basic pharmacotherapeutic profile. Unfortunately, space does not permit a more expanded discussion of clinical trial data or outcome analysis of these agents. Readers may consult many fine reviews for results regarding specific clinical applications.
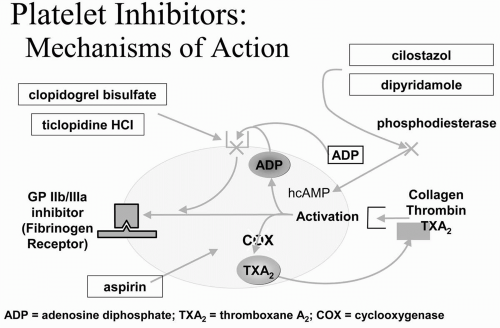 Figure 5-1. Schematic of mechanisms of action of platelet inhibitors. |
Platelet Inhibitors
Platelet inhibitors are basic to the management of patients with vascular disease. Platelet aggregation resulting in plateletrich thrombi is a common pathway causing atherothrombotic events.
The benefits of platelet inhibitors have been clearly recognized. Additional (secondgeneration) platelet inhibitors have been developed and studied in high-risk patients. Oral platelet inhibitors alter platelet function through one of three mechanisms of action (Fig. 5-1) and are commonly used in patients with vascular disease. Blocking the glycoprotein (GP) IIb/IIIa membrane receptor is the most potent form of platelet inhibition; however, this can be achieved only by intravenous infusion.
The GPIIb/IIIa inhibitor agents are restricted to patients undergoing percutaneous coronary intervention.
The GPIIb/IIIa inhibitor agents are restricted to patients undergoing percutaneous coronary intervention.
Aspirin
Aspirin functions by blocking the cyclooxygenase pathway. In so doing, it reduces thromboxane production, thereby reducing the platelet-thrombin interaction. The Antiplatelet Trialist’s Collaboration and, more recently, the Antithrombotics Trialist’s Collaboration, have documented a 25% to 27% risk reduction of major ischemic events in high-risk patients taking aspirin. There is a dose-dependent efficacy observed from the outcome of numerous trials demonstrating greatest treatment benefit in patients receiving 75 mg to 150 mg of aspirin daily. While efficacy improves with lower doses, bleeding complications increase with higher doses.
In patients undergoing vascular reconstruction, pre-operative administration of aspirin has resulted in fewer peri-operative myocardial infarctions (MI), lower mortality, less platelet deposition on endarterectomy sites and prosthetic grafts, and fewer operative strokes in patients undergoing carotid endarterectomy. Moreover, aspirin improves the patency of prosthetic lower extremity bypasses.
Thienopyridines: Ticlopidine and Clopidogrel
The thienopyridines are compounds that, after absorption from the gastrointestinal (GI) tract, are metabolized in the liver. The hepatic metabolite is the active agent that blocks the adenosine diphosphate (ADP) receptor on the platelet membrane. This is a potent form of platelet inhibition. Based upon pharmacodynamic studies, the recommended dose versus platelet effect of ticlopidine and clopidogrel is similar. Ticlopidine is mentioned for historical interest, because its risks of neutropenia and thrombocytopenia have caused most physicians to abandon its use and substitute clopidogrel when this class of compounds is indicated.
The large CAPRIE study compared clopidogrel to aspirin in patients having a recent MI, recent stroke, and chronic peripheral arterial disease (PAD). The overall results demonstrated an 8.7% risk reduction of a major ischemic event in patients randomized to clopidogrel compared to the aspirin group. However, the PAD group enjoyed the greatest benefit, showing a 23.8% risk reduction with clopidogrel. The benefit in the PAD patients carried the overall results in the trial.
Subsequent studies in patients with acute coronary syndromes have shown that combined platelet inhibition with clopidogrel and aspirin reduces major ischemic events in patients treated for acute coronary syndromes and reduces complications of percutaneous coronary intervention. Whether clopidogrel is beneficial in patients undergoing peripheral angioplasty and stenting remains to be established; however, because PAD is established in these patients, management with clopidogrel is justified to achieve the benefit of risk reduction previously mentioned.
Cilostazol
Cilostazol is approved for the improvement of walking distance in patients with intermittent claudication. Cilostazol inhibits phosphodiesterase III, thereby increasing intracellular cyclic AMP (cAMP). In so doing, multiple effects result, including vasodilation, platelet inhibition, inhibition of smooth muscle cells, improved blood flow in animal models, and a reduction in triglycerides and cholesterol.
Patients with intermittent claudication treated with cilostazol have improved walking distances and quality of life. The platelet effect of cilostazol, however, appears modest and does not significantly alter bleeding time in PAD patients. A prospective study evaluating PAD patients taking aspirin, clopidogrel, and cilostazol singly and in combination demonstrated significantly increased bleeding times with aspirin and clopidogrel, but no effect with cilostazol. Cilostazol added to aspirin or clopidogrel or the combination did not change the bleeding time compared to either agent alone or compared to the combination of aspirin plus clopidogrel. Therefore, it appears that cilostazol can be added to other platelet inhibitors without increasing the risk of bleeding.
Anticoagulants
Unfractionated Heparin
Until recently, unfractionated heparin (UFH) was recognized as the most effective anticoagulant. In order to achieve its anticoagulant effect, heparin binds to antithrombin III (ATIII), which converts ATIII from a slow to a rapid inhibitor of fibrin. UFH contains molecular weights ranging from 3,000 to 30,000 daltons. Interestingly, less than one half of administered UFH is responsible for its anticoagulant effect by binding to the ATIII molecule. Secondary anticoagulant effects are achieved by binding to heparin cofactor II, although higher doses of heparin must be administered in order to achieve this effect. Heparin inhibits platelet function and prolongs bleeding time, inhibits vascular smooth muscle cells, and binds to vascular endothelium. These secondary effects may become important after invasive procedures such as arteriography, cardiac catheterization, and angioplasty, both by improving results of these procedures and by increasing their complication rate.
The heparin—ATIII complex inactivates thrombin (factor IIa) and activated factors IX, X, XI, and XII. Evidence is increasing that heparin’s inhibitory effect on coagulation is mediated through the inhibition of thrombin-induced activation of factor V and factor VIII.
The biologic effect (half-life) of heparin does not fit simple first-order kinetics. Higher doses of heparin are accompanied by longer half-lives and vice versa. Therefore, the doseresponse relationship is not linear, and the anticoagulant response increases disproportionately as the dose increases.
Heparin’s action may be prevented by platelets, fibrin, and circulating plasma proteins. Platelets secrete platelet factor 4 (PF4), which actively neutralizes the anticoagulant activity of heparin. Two other plasma proteins, histidine-rich glycoprotein and vitronectin, also neutralize the anticoagulant effect of heparin. Additionally, when factor Xa is bound to platelets, the anticoagulant effect of the heparin-ATIII complex is ineffective. Although heparin has varying effects on the plasminogen-plasmin enzyme system, the overall effect on endogenous fibrinolytic activity is small, and heparin most likely neither enhances nor inhibits endogenous fibrinolysis.
Clinical trials have demonstrated that continuous intravenous heparin is safer and more effective than intermittent, bolus intravenous infusion for the treatment of thrombotic disorders. Additionally, therapeutic anticoagulation with heparin is defined as increasing the activated partial thromboplastin time (aPTT) greater than 1.5 times baseline. Failing to achieve this level continuously from the onset of treatment of venous thromboembolic disorders significantly increases recurrence rates.
Heparin-induced thrombocytopenia (HIT) is a well recognized and feared complication of heparin that is usually observed 5 to 10 days after heparin use has begun. HIT is an antigen-antibody immunologic response that is not dose related. It is caused by heparin-induced antiplatelet antibodies
leading to platelet aggregation, thrombocytopenia, and the subsequent thromboembolic complications.
leading to platelet aggregation, thrombocytopenia, and the subsequent thromboembolic complications.
Table 5-1 Comparison of Available LMWHs | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
HIT can occur rapidly if the patient has existing antibodies at the time heparin is administered. Platelet counts should be monitored in all patients receiving heparin, regardless of the route of administration or the dose prescribed. When HIT is diagnosed, it should be treated.
Low-Molecular-Weight Heparin
Four low-molecular-weight heparins (LMWHs) have been approved for use in the United States: ardeparin, dalteparin, enoxaparin, and tinzaparin. However, only dalteparin and enoxaparin are currently marketed. Several other agents are available in Canada and European countries, including bemiparin, certoparin, fraxiparin, nadroparin, and reviparin. While the mechanism of action is similar between agents, all LMWHs are not the same. The dosing and anticoagulant activity vary between the agents, and thus familiarity with the available agents is required for their use.
LMWHs are formed by the enzymatic or chemical fragmentation of porcine UFH. A mixture of glycosaminoglycan molecules, which is considerably smaller than UFH (approximately 5000 daltons), is produced. Because of their small size, the pharmacology and pharmacokinetics of LMWH are distinct from UFH. Similar to UFH, LMWH binds to antithrombin (AT) via a specific pentasaccharide sequence. This binding induces a conformational change within AT and accelerates the binding and clearance of activated factors X (Xa) and thrombin (IIa). However, because most of the LMWH molecules are approximately 15 saccharide units in length, their ability to bind to factor IIa is limited; therefore, factor Xa inhibition is the predominant anticoagulant effect (Table 5-1).
There are many considerations when deciding between the use of LMWH or UFH for similar clinical indications (Table 5-2). When administered by subcutaneous injection, the bioavailability is approximately 90%, compared to 30% for UFH. In part, this is due to reduced binding to the endothelium, plasma proteins, albumin, macrophages, and platelets. Reduced nonspecific binding and a longer plasma half-life allow for once-daily or twice-daily administration by subcutaneous injection for most clinical indications (Table 5-3). The ease of administration and the potential for outpatient therapy make LMWH a favorable choice for treatment of DVT, DVT prophylaxis in the hospital setting, and prolonged DVT prophylaxis following orthopedic surgery.
In most clinical settings no monitoring is required when using LMWH. Monitoring may be helpful or necessary in patients with hepatic or renal insufficiency as well as in pediatric, pregnant, obese, or very thin patients. However, unlike UFH, LMWHs do not prolong the aPTT. Therefore, when it is indicated, the preferred method of monitoring is a chromogenic anti-Xa assay using an LMWH control. A chromogenic anti-Xa activity level performed 4 hours following subcutaneous injection should be in the range from 0.5 to 1.1 for a therapeutic dose and 0.2 to 0.3 for a prophylactic dose.
LMWHs are cleared by renal excretion. In patients with mild (CrCL 50 to 80 ml/min) or moderate (CrCl 30 to 50 ml/min) renal insufficiency, no dose adjustment is usually required. However, when used in patients with severe renal insufficiency (CrCL <30 ml/ min), dose adjustment or monitoring may be necessary (Table 5-4). There is no agent that reliably reverses the anticoagulant effect of LMWH. Protamine does not neutralize LMWH to the same extent as UFH; the binding for LMWH is reduced. Although protamine remains a recommended antidote for LMWH, the effect on clinically significant bleeding may not prove beneficial. LMWH should be used with caution in patients with an increased risk for bleeding and those with relative contraindications to anticoagulation (Table 5-5).
Table 5-2 Advantages and Disadvantages of LMWH Compared to UFH1 | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||



