Chapter 7 Pharmacology of Antithrombotic Drugs
Atherothrombotic disease and atherosclerotic plaque rupture is the leading cause of death worldwide. Its prevalence among adults in the United States is estimated at over 81 million, with costs exceeding $503 billion annually.1 Thrombus and clot formation involved in atherothrombotic disease develop as a complex and dynamic interaction between platelets, blood vessel wall, and coagulation cascades. Increased understanding of the mechanism of these interactions has provided for the development of novel drugs. Antiplatelet drugs have come to the forefront in managing atherothrombotic disease, owing in large part to platelets’ involvement in the initiation and propagation of thrombus. Our understanding of platelet function has expanded from a rudimentary knowledge of aspirin as a cyclooxygenase (COX) inhibitor within the arachidonic acid pathways, to a more complex picture of multiple receptor-modulating agents including thienopyridines, glycoprotein (GP)IIb/IIIa receptor antagonists, von Willebrand factor (vWF)-GPIb/IX, and collagen-GPVI inhibitors. Despite advances with newer inhibitors and combinations, treatment failures persist, necessitating development of new antiplatelet agents.
Platelets, Thrombosis, Coagulation, and Atherothrombotic Vascular Disease
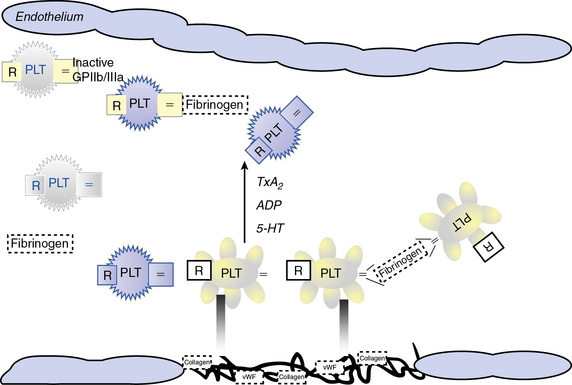
Atherosclerotic plaque rupture and endothelial cell (EC) disruption lead to platelet activation and formation of occlusive thrombi, triggering acute ischemic events in patients with atherothrombotic disease. Platelet activation and aggregation involve multiple signaling molecules and their receptors. Initially, platelets adhere to the subendothelial proteins vWF and collagen), which are exposed at sites of vascular injury. Adenosine diphosphate (ADP), thromboxane A2 (TxA2), serotonin, collagen, and thrombin activate platelets through unique intracellular signaling pathways, resulting in further platelet activation and secretion of mediators, thus further amplifying and sustaining the initial platelet response.2 Adenosine diphosphate, serotonin, and calcium are released by activated platelets via degranulation; thromboxane from arachidonic acid; and thrombin from activated coagulation cascade pathways.3 Activation of platelets occurs through binding of their primary blood-soluble agonists to their respective platelet receptors: ADP binds P2Y1 and P2Y12, thrombin binds to protease- activated receptor 1 (PAR1) and PAR4, and thromboxane binds to the thromboxane/prostanoid (TP) receptor.4 The final common pathway for all autocrine and paracrine activation signals is GPIIb/IIIa activation, which mediates fibrinogen and vWF binding to platelets and contributes to platelet aggregation. Thus, in both physiological hemostasis and pathological states, platelets are recruited to form a platelet-fibrin thrombus.3,4 Various classes of antiplatelet drugs act synergistically through complementary yet independent mechanisms, preventing platelet aggregation and thus acute thrombus formation. Currently available drugs and those under investigation target the thromboxane-induced (aspirin, sulfinpyrazone, indobufen, and triflusal) and ADP-induced (ticlopidine, clopidogrel, prasugrel, ticagrelor, cangrelor and elinogrel) pathways of platelet activation and their final common pathway of GPIIb/IIIa-induced (abciximab, eptifibatide, and tirofiban) platelet aggregation.4,5 The processes of platelet adhesion, activation, and aggregation, along with the targets of platelet-inhibiting drugs, are shown in Figure 7-1. Antiplatelet drugs, in addition to inhibiting acute arterial thrombosis, interfere with the physiological role of platelets in hemostasis. Thus the range of adverse effects, particularly bleeding, is a major factor in evaluating the utility of available and upcoming antiplatelet drugs and their combination regimens. Coagulation cascades are intimately activated through atherosclerotic plaque rupture and platelet activation. Targets of drug therapy to regulate the effects of thrombus formation and propagation are accomplished through oral anticoagulation (warfarin); thrombin inhibitors, both direct and indirect (heparin, low-molecular-weight heparin [LMWH], fondaparinux, hirudins, bivalirudin, argatroban, ximelagatran, dabigatran, etexilate, rivaroxaban, apixaban, DU-176b, LY517717, betrixaban, and YM150); factor IX inhibitors; and factor Xa inhibitors.
Pharmacology of Platelet Inhibitors
Platelets circulate in blood with their activation inhibited by both nitric oxide (NO) and prostaglandin I2 released from ECs.6,7 Activated platelets prevent bleeding by catalyzing the formation of stable blood clot in conjunction with activated coagulation pathways. In the initiation phase of primary hemostasis, platelets roll, adhere, and spread along the exposed collagen matrix of injured blood vessels to form an activated platelet monolayer.8 During the rolling phase, platelet adhesion and tethering is mediated by the platelet GPIb/V/IX receptor complex and vWF, which itself is bound to collagen (see Fig. 7-1). Additional tethering is accomplished between the GPVI and GPIa proteins directly with collagen at sites of vascular injury.6–8 The binding of GPIb/V/IX to vWF has a fast off rate insufficient to mediate stable adhesion, but instead, able to maintain platelets in close contact with the endothelial surface. Platelet activation stimulates high-affinity integrins to form stable adhesion complexes.
Blood flows with greater velocity in the center of the vessel than near the wall, thereby generating shear forces between adjacent layers of fluid. In conditions of high shear, such as those of small arteries, arterioles, and stenosed arteries, the tethering process is integral in the mechanisms of platelet adhesion. von Willebrand factor binds to collagen within the extracellular matrix (ECM) and to platelet receptors (GPIb/V/IX and GPIIb/IIIa [αIIβ3 integrin]).8 Binding of ECM collagen triggers intracellular signals that shift platelet integrins to a higher-affinity state and induce release of the secondary mediators ADP and TxA2. Both ADP and TxA2, along with thrombin produced from the coagulation cascade, synergistically induce full platelet activation. Upon platelet activation, arachidonic acid is liberated from membrane phospholipids by phospholipase A2 and C, thereby producing TxA2. Aspirin and other agents, such as sulfinpyrazone, indobufen, and triflusal, act to inhibit enzymes within the arachidonic acid cascade, thereby limiting production of TxA2. Adenosine diphosphate binds to P2Y1 and P2Y12 surface platelet receptors, which are targets of clopidogrel, prasugrel, and ticagrelor.
Thrombin is produced at the surface of activated platelets by tissue factor and is responsible for generating fibrin from fibrinogen, which contributes to formation of the hemostatic plug and platelet thrombus growth. Thrombin also directly activates platelets through stimulation of the PAR1.7 Both direct and indirect inhibitors of thrombin inhibit thrombin and affect thrombin activation, respectively. Release of ADP and TxA2 from adherent platelets contributes to recruitment of circulating platelets, thereby inducing a change in platelet shape, increased expression of proinflammatory molecules (P-selectin, CD40 ligand), expression of platelet procoagulant activity, and conversion of the GPIIb/IIIa receptor into an active form, leading to pathological thrombosis.6–8 Activation of GPIIb/IIIa (αIIβ3 integrin) mediates platelet aggregation and spreading on the exposed ECM of the injured vessel wall by means of fibrinogen bridges.8
Fibrinogen bridges activated platelets and contributes to thrombus stabilization.8 Activation of platelets results in a conformational change in the αIIβ3 integrin (IIb/IIIa) receptor, enabling it to bind fibrinogen-enhancing cross-links to adjacent platelets, resulting in aggregation and formation of a platelet plug. Simultaneous activation of the coagulation system results in thrombin generation and fibrin clot formation, which further stabilizes the platelet plug. Abciximab, eptifibatide, and tirofiban inhibit GPIIb/IIIa, thereby inhibiting platelet aggregation.
Cyclooxygenase Inhibitors and the Arachidonic Acid Cascade
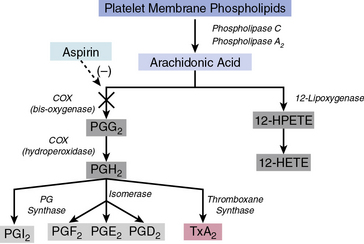
Arachidonic acid is liberated from membrane phospholipids by phospholipase A2 and C upon platelet stimulation9 (Fig. 7-2). Prostaglandin (PG) H-synthase catalyzes the conversion of arachidonic acid to PGG2 and PGH2.10 Prostaglandin-synthase possesses two catalytic sites, a COX site, responsible for the formation of PGG2, and a hydroperoxidase site, which reduces the 15-hydroperoxyl group of PGG2 to produce PGH2.11 Subsequent enzymatic catalyzation of PGH2 generates PGs, D2, E2, F2α, I2, and TxA2.11 Aspirin irreversibly binds and inhibits the COX site by acetylating the hydroxyl group of a serine residue at position 529 (Ser 529) without affecting the hydroperoxidase activity of the enzyme, thus inhibiting production of PGG2 and therefore PGH2 circumventing TxA2 production.12
Aspirin
Aspirin produces dose-dependent inhibition of platelet COX activity after a single oral dose. A single dose of 100 mg effectively suppresses biosynthesis of TxA2 within several minutes of administration via acetylation of platelet COX in the presystemic circulation.13 Owing to aspirin’s irreversible inhibition of COX and the inability of platelets to synthesize new proteins, aspirin’s effect is maintained for the lifespan of the platelet (7-10 days). Cyclooxygenase activity returns only as new platelets are generated.
Aspirin reduces acute coronary and cerebrovascular events such as unstable angina, myocardial infarction (MI), sudden cardiac death, and stroke.13 Its utility is enhanced by its modest cost and nominal side effects. Although aspirin effectively reduces platelet secretion and aggregation, it is a relatively weak platelet inhibitor. The inhibitory effects of aspirin are pronounced when using relatively weak platelet agonists, but less so against stronger agonists like thrombin that can induce platelet activation in the absence of TxA2. Importantly, the majority of platelet responses remain unaffected by aspirin treatment. Aspirin does not inhibit shear stress–induced platelet activation and platelet adhesion. In addition to its antiplatelet properties, aspirin also exerts antiinflammatory effects.14 In a meta-analysis of antiplatelet therapy studies in patients with acute coronary syndrome (ACS), administration of several doses of aspirin (from 75 mg/day up to 150 mg/day) significantly decreased the overall risk of nonfatal MI, nonfatal stroke, and death rates.15 Several studies, however, have demonstrated that aspirin monotherapy is inadequate because of high intraindividual variability to aspirin response, as well as increased aspirin resistance, especially observed in patients with diabetes mellitus.16
Nonsteroidal antiinflammatory drugs
There are several nonsteroidal antiinflammatory drugs (NSAIDs) that act as competitive reversible inhibitors of PGH-synthase. Sulfinpyrazone, indobufen, and triflusal are several of the drugs in this class evaluated for their antithrombotic activity in randomized clinical trials. The active sulfide metabolite of sulfinpyrazone administered in the highest dose allowable (200 mg four times a day) inhibits only 60% of platelet COX activity, with results suggesting no significant clinical efficacy.17 The clinical, biochemical, and functional effects of the more effective inhibitor, indobufen, are similar to those of aspirin. An oral dose of indobufen 200 mg twice daily inhibits 95% of platelet TxA2 synthesis.18 Triflusal, a derivative of salicylic acid, is also able to inhibit platelet COX, but only after conversion to a longer-lasting metabolite.19 None of the three are currently approved for use as antiplatelet drugs in the United States.
Adenosine Diphosphate, Purinergic Receptors, and Thienopyridine Inhibitors
Adenosine diphosphate is a key mediator in activating platelet aggregation and thrombus formation. Inhibiting the effects of ADP activity has lead to development of numerous P2Y12 receptor–targeting antiplatelet drugs. Adenosine diphosphate is released from dense granules of activated platelets, providing a soluble positive feedback mediator binding to the receptors P2Y1 and P2Y12. Both P2Y1 and P2Y12 are platelet surface-bound purinoreceptors belonging to the G protein–coupled receptor (GPCR) class, with P2Y1 being coupled to Gq and P2Y12 to Gi. Adenosine diphosphate binding to both P2Y1 and P2Y12 receptors activates distinct intracellular signaling pathways.20 Binding of ADP to the P2Y1 receptor and its Gq protein mobilizes intracellular calcium, triggering a change in the platelet shape and rapid, reversible aggregation.21 Adeosine diphosphate binding through the P2Y12 receptor and its Gi protein results in reduced levels of cyclic adenosine monophosphate (cAMP), resulting in amplification of the platelet response, stabilization of resulting aggregates, and secretion of further mediators from platelet granules.22 Binding of ADP to both P2Y1 and P2Y12 purinoreceptors is necessary for normal ADP-induced aggregation. P2Y12 is considered the major platelet ADP receptor and, since it is more restricted in its expression throughout cell lines, has become an attractive therapeutic target for antithrombotic agents.20
Thienopyridine platelet P2Y12 receptor antagonists
The thienopyridine class of antiplatelets (ticlopidine, clopidogrel, and prasugrel) selectively and irreversibly inhibit the P2Y12 purinoreceptor throughout the life of the platelet. Clopidogrel is the dominant member within the class and provides for modest platelet inhibition, delayed onset of action, and significant interpatient variability, including nonresponsiveness to the drug, which necessitated the search for more potent and stable alternatives.23 Ticlopidine has been eclipsed because of its adverse hematological side effects, including neutropenia and thrombotic thrombocytopenic purpura.24 The opposite, however, is true of third-generation thienopyridines, namely prasugrel.
Oral thienopyridines are prodrugs that require conversion into their active metabolites by hepatic cytochrome P450 (CYP) enzymes (CYP3A4 isozyme). Whereas clopidogrel requires esterase inactivation and a two-step CYP-dependent activation, prasugrel requires only one reaction to yield its active metabolite.25 This difference in metabolism translates into different patient responses and drug interactions. Genetic variations of P450 (CYP) enzymes affect clopidogrel’s active metabolite formation, resulting in lower platelet inhibition and, most importantly, a higher rate of major adverse cardiovascular events. Prasugrel’s pharmacology, however, is not affected by CYP polymorphisms and provides a stable platform for antiplatelet therapy. Prasugrel has a faster onset of action and a tenfold higher potency than clopidogrel.26
Clopidogrel
Clopidogrel is metabolized by CYP P450. Its active metabolite irreversibly binds to the platelet P2Y12 receptor, thus inhibiting the effect of ADP on platelets. As a result, GPIIb/IIIa receptors have decreased activation, thereby resulting in reduced platelet function. The Clopidogrel versus Aspirin in Patients at Risk of Ischemic Events (CAPRIE) study, a randomized trial that included patients with ischemic stroke, MI, or symptomatic atherosclerotic peripheral artery disease, showed that clopidogrel-treated patients had an 8.7% relative risk reduction for acute MI, stroke, or vascular death, compared to those patients treated with aspirin.27 The Clopidogrel in Unstable Angina to Prevent Recurrent Events (CURE) trial was the first study establishing the significance of dual antiplatelet therapy (acetylsalicylic acid [ASA] plus clopidogrel) in patients with ACS28 and involved a population of 12,562 patients presenting with non-ST-elevation ACS who were randomized to receive either a combination of ASA (75-325 mg/day) and clopidogrel (300 mg loading dose, followed by 75 mg/day) or ASA and placebo for 3 to 12 months. At 12 months, a lower incidence of MI, stroke, or cardiovascular death was observed in the clopidogrel plus ASA group compared to the placebo group; however, the risk of major bleeding was increased among patients treated with clopidogrel plus ASA.
Similar results were reported by the COMMIT study.29 Patients received either a combination of clopidogrel (75 mg/day) and ASA (162 mg/day) or placebo and ASA for 28 days or until hospital discharge. There was a 9% relative risk reduction in the composite endpoint of stroke, vascular reinfarction, or death and a 7% relative risk reduction for death in the group receiving dual antiplatelet therapy. In the CREDO study, which was the first to evaluate the significance of dual antiplatelet therapy pre- and post-percutaneous coronary intervention (PCI), patients undergoing PCI received either a 300-mg loading dose of clopidogrel 3 to 24 hours before the procedure and then 75 mg/day for 12 months, or 75 mg/day for 30 days after the procedure without a loading dose.30 All patients also received a 325 mg/day dose of ASA during the 12-month follow-up period. A 27% relative risk reduction for death, MI, or stroke was observed in the group receiving long-term dual antiplatelet therapy. Given the established antiplatelet effect of clopidogrel, the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization Management and Avoidance (CHARISMA) study investigated the potential benefit from 28-month dual antiplatelet therapy (75 mg/day clopidogrel and 75-162 mg/day ASA) in patients 45 years of age or older who experienced one of the following conditions: multiple atherothrombotic risk factors, documented coronary disease, documented cerebrovascular disease, or documented symptomatic peripheral artery disease.31 The control group received ASA and placebo. No benefit was observed in the primary endpoint of MI, stroke, or cardiovascular death in the dual antiplatelet therapy group. By contrast, asymptomatic patients of the group with risk factors but without clinical evidence of CAD had a higher risk of bleeding.32 A further analysis in the subgroup of patients with documented atherothrombotic cardiovascular disease showed a significant risk reduction for new MI, stroke, or cardiovascular death in those patients compared with controls.33
Prasugrel
Prasugrel is a P2Y12 inhibitor acting similarly to clopidogrel. Prasugrel is rapidly converted to its active metabolite by the P450 cytochrome and has higher bioavailability than clopidogrel.26 Recently, a 60-mg loading dose of prasugrel achieved high platelet inhibition both in healthy subjects and in patients scheduled for PCI, whereas healthy clopidogrel poor-responders achieved satisfactory platelet inhibition of up to 80% after prasugrel administration.26 The beneficial effect of prasugrel was also established in the TRITON-TIMI 38 study.34 Patients scheduled for PCI were randomized to receive either clopidogrel (300-mg loading dose and 75 mg/day afterwards) or prasugrel (60-mg loading dose and 10 mg/day afterwards). All patients also received ASA, and about half the patients from each group were treated with a GPIIb/IIIa inhibitor as well. The prasugrel group demonstrated a significant risk reduction for the composite primary endpoint of death, nonfatal MI, and nonfatal stroke. In addition, risk was significantly lower in the diabetes mellitus patient subgroup34; however, incidence of major hemorrhagic events was more frequent in the prasugrel group, but even when this parameter was added to the study’s primary endpoints, the net clinical benefit findings still favored prasugrel compared to clopidogrel. Subgroup analysis demonstrated a higher rate of major bleeding in those with body weight of less than 60 kg, history of stroke or transient ischemic attack, and age older than 75 years.34
Non-thienopyridine platelet P2Y12 receptor antagonists
Novel non-thienopyridine platelet P2Y12 receptor antagonists include ticagrelor, cangrelor, and elinogrel, which are direct and reversible P2Y12 antagonists with rapid onsets and short durations of action. Ticagrelor is highly selective and very specific for the P2Y12 receptor, and it exhibits a greater, more consistent inhibition of platelet aggregation than clopidogrel.35 Ticagrelor is administered orally and does not require metabolic activation, providing a rapid onset peaking within 2 to 4 hours of dosing. The metabolism of ticagrelor yields an active molecule (AR-C124910XX) that has similar P2Y12-blocking activity as its parent molecule. Ticagrelor’s plasma half-life is approximately 12 hours, which corresponds to twice-daily dosing and a 1- to 2-day restoration of normal platelet-mediated hemostasis upon discontinuation. These pharmacokinetics are in contrast to clopidogrel and prasugrel, which require discontinuation approximately 5 days before restoration of normal platelet-mediated hemostasis is achieved. Ticagrelor’s potential advantage in plasma half-life also carries the risk of increased thrombotic events if patients miss a dose.36
Multiple trials have demonstrated the benefits of ticagrelor in clinical practice. The Dose Confirmation Study Assessing Antiplatelet Effects of AZD6140 vs. Clopidogrel in Non-ST-Segment Elevation Myocardial Infarction (DISPERSE-2) trial showed no difference in major bleeding or MI, with an increase in minor bleeding at higher doses of ticagrelor in 990 patients with non-ST-segment elevation ACS.37 In the PLATO (the Study of Platelet Inhibition and Patient Outcomes) trial comparing ticagrelor and clopidogrel with respect to their efficacy in preventing cardiovascular events and safety showed ticagrelor significantly reduced the rate of death from vascular causes, MI, or stroke, without an increase in the rate of overall major bleeding compared to clopidogrel in 18,624 patients with ACS.38 Ticagrelor is the first investigational antiplatelet drug to demonstrate a reduction in cardiovascular death when compared to clopidogrel in patients with ACS.
Finally, research has shown ticagrelor to produce platelet inhibition regardless of genotypic variations in the three genes that had been associated with variability to clopidogrel in platelet inhibition.39 All these trials underline the potential for ticagrelor to achieve a rapid and sustained antiplatelet effect that could be reversed and could overcome nonresponsiveness and interpatient variability to clopidogrel, thus addressing the main limitations of clopidogrel therapy.24 Nonetheless, its adverse effects (e.g., dyspnea, bradycardia) and weight-based dosing require further investigation before ticagrelor may advance toward routine use in antiplatelet therapy.38
Role of Integrin Receptors in Platelet Function
Abciximab
Abciximab is a large chimeric monoclonal antibody Fab fragment with high affinity for the GPIIb/IIIa receptor.40 Abciximab is the largest agent with binding sites located on the β-chain of the GPIIb/IIIa receptor which, because of its large size, causes a steric hindrance to ligand access.
The efficacy and safety of abciximab in patients undergoing PCI has been evaluated in several trials, including EPIC, EPILOG, and EPISTENT.41–43 The Intracoronary Stenting and Antithrombotic Regimen–Rapid Early Action for Coronary Treatment (ISAR-REACT) trial demonstrated that the rate of death, MI, and urgent revascularization at 30 days was low and comparable between patients undergoing elective PCI after pretreatment with clopidogrel 600 mg and allocated to abciximab vs. placebo.44 Rates of major bleeding were similar between groups, although abciximab was associated with a significantly higher rate of thrombocytopenia. ISAR-REACT-2 evaluated the same abciximab and clopidogrel treatment regimens in high-risk patients with non-ST-segment elevation ACS undergoing PCI.45 Death, MI, and urgent revascularization at 30 days occurred significantly less frequently with abciximab compared to placebo; however, the treatment benefit of abciximab was confined to patients with elevated troponin levels. Rates of major bleeding were similar between groups. Overall, the findings suggest that in the modern era of interventional cardiology using high clopidogrel dosing regimens, GPIIb/IIIa inhibition should be reserved only for high-risk ACS patients with positive cardiac markers.
Eptifibatide
Eptifibatide is a competitive antagonist of the GPIIb/IIIa receptor. It is a synthetic small-molecule inhibitor that fits directly into the Arg-Gly-Asp binding pocket of the GPIIb/IIIa receptor, directly competing with the binding of ligands such as fibrinogen and vWF.46 Eptifibatide rapidly dissociates from its receptor, is cleared by the kidney largely as active drug, and has a plasma half-life of approximately 1.5 to 2.5 hours. The return of hemostatic platelet function is largely dependent on clearance of the drug from plasma. Cessation of drug infusion restores platelet function and, in patients with normal renal function, normal hemostasis returns within 15 to 30 minutes after drug discontinuation. Unlike abciximab, however, the platelet inhibitory effect of eptifibatide is not significantly influenced by platelet transfusion.46
Eptifibatide has demonstrated efficacy and safety in patients with non-ST-segment elevation ACS or undergoing PCI in a number of randomized clinical trials. Most recently, the Early Glycoprotein IIb/IIIa Inhibition in Non-ST-Segment Elevation Acute Coronary Syndrome (EARLY ACS) trial demonstrated that early administration of eptifibatide vs. provisional eptifibatide after angiography (delayed eptifibatide) resulted in similar 30-day rates of death, MI, urgent revascularization, or thrombotic complications during PCI in patients with non-ST-segment elevation ACS undergoing invasive management.47 Major and minor bleeding rates were significantly higher with early eptifibatide vs. delayed eptifibatide. Overall, these findings do not support the use of upstream compared with ad hoc GPIIb/IIIa inhibition in ACS patients undergoing PCI.
Tirofiban
Tirofiban is a tyrosine-derived nonpeptide inhibitor associated with rapid onset and short duration of action, with a plasma half-life of approximately 2 hours. Tirofiban, like eptifibatide, is a competitive inhibitor of the GPIIb/IIIa receptor that has high specificity but relatively low affinity.46 Tirofiban is excreted by the kidney, predominantly as unchanged drug; it rapidly dissociates from the receptor and has a biological half-life of 1.5 to 2.5 hours. Restoration of hemostasis is best achieved by discontinuing the drug infusion. Efficacy and safety of tirofiban in PCI patients has been investigated in several trials.
Platelet Adhesion
von Willebrand factor is present in plasma, platelets, and vascular subendothelium, and is synthesized and stored by megakaryocytes and ECs.48 von Willebrand factor can be released into the circulation by ECs upon activation by vasopressin (desmopressin/DDAVP [1-deamino-8-D-arginine-vasopressin]) or thrombin, for example.48 von Willebrand factor serves two important functions in the hemostatic response of platelets: initially through platelet/platelet binding (GPIba/vWF A1 domain), and subsequently through collagen/platelet binding (GPIba/vWF A3 domain). The vWF A1 domain specifically serves to assist with platelet aggregation by binding to platelet GPIba in the GPIb/V/IX complex for platelet/platelet adhesion, especially under conditions of high shear stress. von Willebrand factor additionally promotes platelet adhesion by binding to collagen in exposed vascular subendothelium via the vWF A3 domain. The adhesion function of the GPIb/V/IX complex that interacts with vWF resides in the GPIba chain; mutations within this segment may alter its affinity for the vWF A1 domain. 49
Von willebrand factor–GPIP/IX inhibitors
A number of vWF-GPIb/IX inhibitors, including aurin tricarboxylic acid, peptide fragments from vWF A1 domain, and a soluble GPIb-immunoglobulin (Ig)G, have been examined for inhibiting vWF and platelet interactions.50 Glycoprotein-290 is a recombinant protein consisting of a 290 amino acids sequence of the N-terminal of human GPIba, with two gain-of-function mutations that increase the affinity for the vWF A1 domain. The in vivo antithrombotic and antihemostatic effects have been evaluated with good GPIba/vWF inhibition, which could be reversed with a currently approved (DDAVP) treatment regimen. A lower clopidogrel dose combined with GPIba/vWF inhibition could theoretically have improved efficacy with less bleeding risk.
AJvW-2 is a murine monoclonal antibody to human vWF A1 domain that blocks the GPIba/vWF interaction and has demonstrated antithrombotic activity in animal models.51 In order to reduce immunological response, AJvW-2 was humanized and converted from an IgG1 to an IgG4.52 This humanized antibody (AJW200) exhibited similar inhibition of in vitro vWF-mediated platelet activation to AJvW-2. In human volunteer studies, AJW200 demonstrated no clinically significant adverse events or immunogenicity. Ristocetin cofactor (Ri:CoF) assays showed a significant reduction at 1 hour post infusion compared with baseline that lasted for up to 12 hours. The template bleeding time was not significantly prolonged at any time or dose of AJW200. Platelet function as measured by the PFA-100 was reduced by up to 3 to 6 hours at the lower dose and 12 hours at the highest dose administered.
ARC1779 is an aptamer that blocks the GPIba/vWF A1 domain interaction. Aptamers are nucleic acid molecules with high affinity and specificity for a selected target molecule, discovered through in vitro selection on the basis of their ability to fold into unique three-dimensional structures that promote binding to that target.53 ARC1779 is a modified deoxyribonucleic acid/ribonucleic acid (DNA/RNA) oligonucleotide composed of hybrid terminal ends to minimize endonuclease and exonuclease digestion, with nucleotide segments designed to enhance affinity for vWF. ARC1779 has demonstrated efficacy comparable or superior to that of previously published dosing regimens of abciximab with respect to protection from thrombus formation and average time to occlusion. ARC1779 was evaluated in a randomized double-blind placebo-controlled study that demonstrated it was well tolerated, and no bleeding was observed.54 An S-nitroso derivative of a mutated fragment of the A1 domain (S-nitroso-AR545C) was shown to inhibit effectively arterial thrombosis in the carotid artery.55 In unpublished observations, it has also been reported that targeting the vWF A1 domain with the recombinant nanobody ALX-0081, a novel class of antibody therapeutics, resulted in inhibition of vWF-mediated platelet activation, providing novel options for future therapies.
Collagen-GPVI inhibitors
Glycoprotein VI is also expressed on the surface of platelets. Signaling by GPVI is via an immunoreceptor tyrosine activation motif (ITAM) promoting phosphorylation and initiating the syk signaling cascade. Syk activation results in activation of integrin-induced platelet aggregation, release of ADP and thromboxane, and procoagulant activity.56 Rat monoclonal antibody (JAQ1) to mouse GPVI results in inhibition of collagen-induced aggregation.57 In animal models, JAQ1 caused mild thrombocytopenia and resulted in a 34% decrease in platelet counts within 24 hours of treatment, which returned to normal levels within 3 days of treatment. Platelets showed no indication of activation or change in surface protein expression. A single dose of JAQ1 resulted in 14 days of inhibition of ex vivo collagen-induced aggregation. Further evaluation of in vivo response determined that binding of antibody to the platelet GPVI resulted in depletion of platelet GPVI. Collagen-induced adhesion was significantly reduced in these GPVI-depleted platelets. Bleeding times of JAQ1-treated mice were significantly elevated over control mice (330 seconds vs. 158 seconds), but less than that seen following inhibition of GPIIb/IIIa (330 seconds vs. > 600 seconds).
Pharmacology of Antithrombotics and Thrombin Inhibitors
Overview of Coagulation
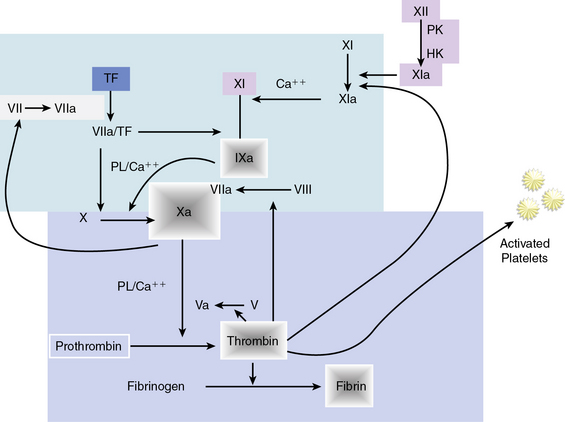
Hemostasis is accomplished by a complex sequence of interactions among platelets, endothelium, and multiple circulating and membrane-bound coagulation factors. As shown in Figure 7-3, the coagulation cascade typically has two intersecting pathways. The intrinsic pathway is initiated with factor XII and involves a cascade of enzymatic reactions that activate factors XI, IX, and VII. In the intrinsic pathway, all factors leading to fibrin clot formation are intrinsic to the circulating plasma, and no surface is required to initiate the process. The extrinsic pathway, however, requires exposure of tissue factor on the surface of the injured vessel wall to initiate the cascade, beginning with factor VII. The two arms of the coagulation cascade merge to a common pathway at factor X, which activates factors II (prothrombin) and I (fibrinogen). The formation of clot is dependent upon the proteolytic conversion of fibrinogen to fibrin.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


