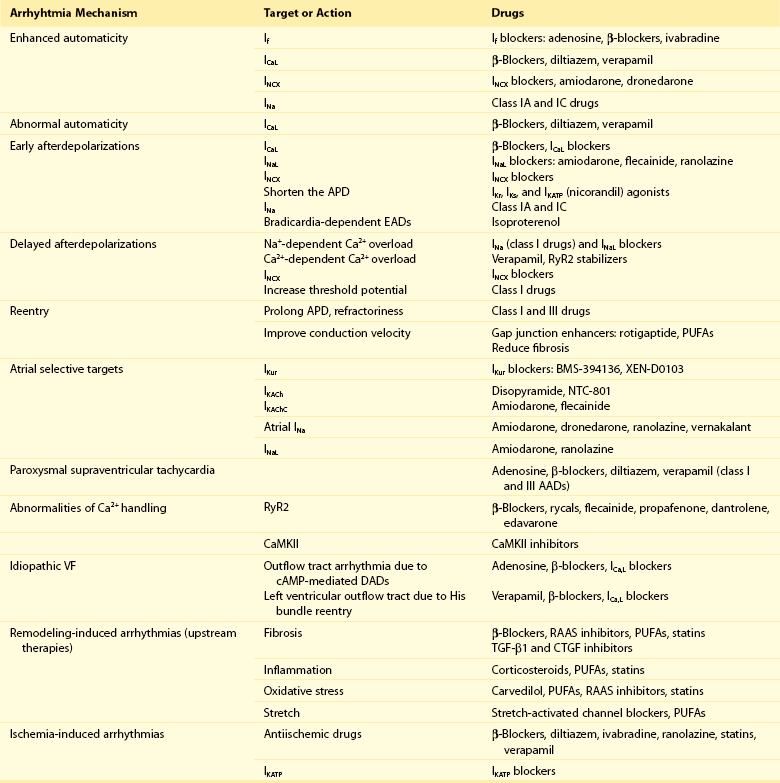
54 Antiarrhythmic Drugs: Introduction Na+ Channel Blockers: Class I AADs K+ Channel Blockers: Class III Antiarrhythmic Effects Calcium Channel Blockers: Class IV AADS Gap-Junction Coupling Enhancers Stretch-Activated Ion Channels Modulation of Ion Channel Trafficking Targeting Intracellular Calcium Handling Pharmacologic Treatment of Inherited Cardiac Arrhythmia Syndromes Target Cardiac Remodeling: Upstream Therapies Unresolved Questions and Future Strategies Cardiac arrhythmias represent a problem in current clinical practice; they are a major cause of morbidity and mortality and are an increasing economic burden for health care systems.1 Treatment of cardiac arrhythmias with antiarrhythmic drugs (AADs) has two main objectives: relieve symptoms and complications (improve quality of life) and reduce mortality directly related to the arrhythmia. A basic principle in pharmacology is that the best treatment is targeted specifically to disease mechanisms. However, in many patients the ultimate underlying mechanisms of the arrhythmia remain incompletely understood. Thus, the choice of a given AAD is empiric and based on the characteristics of the arrhythmia, the pharmacologic properties of the AAD, and, above all, its safety profile. Moreover, triggers and arrhythmogenic substrates can vary among patients with the same arrhythmia depending on the underlying structural heart disease (i.e., coronary artery disease [CAD], heart failure [HF], left ventricular [LV] hypertrophy, hypertension). This variation could explain why AADs produce widely divergent effects on different patients ranging from termination of the arrhythmia to inefficacy, to exacerbation of the treated arrhythmias or generation of entirely new ones (proarrhythmia). Until recently, arrhythmias were primarily considered a purely electrophysiological problem, and AADs mainly target cardiac Na+, Ca2+, and K+ ion channels (Figure 54-1). They bind to specific receptor sites within the channel drug affinity being strongly modulated by the channel state in a time- and voltage-dependent manner.2 In addition, some AADs modulate the autonomic tone, primarily by antagonizing β1-adrenoceptors (β-blockers) or muscarinic receptors (atropine, disopyramide) or by stimulating adenosine A1 receptors (adenosine). Figure 54-1 Ionic currents involved in shaping of human atrial (A) and ventricular (B) action potentials. The initial upstroke of the action potential is due to the activation of the peak inward Na+ current (INa). Repolarization is determined by the balance between inward-depolarizing L-type Ca2+ (ICaL) and late Na+ (INaL) currents and outward-repolarizing K+ currents, including the rapidly activating and inactivating transient current (Ito1), ultrarapid (IKur), rapid (IKr), and slow (IKs) components of the delayed rectifier, the inward rectifier current (IK1) and the ligand-gated currents activated by a decrease in the intracellular concentration of adenosine triphosphate (IKATP) or activated by acetylcholine (IKAch) or adenosine (IKAdo). Some currents are exclusive to the atria (IKur) or more important (Ito1, IKACh) in the atria than in the ventricles and vice versa (IKr). Arrows indicate the ion movement direction. Conventional AADs are most commonly grouped according to the Vaughan-Williams classification in four different groups (Table 54-1): Na+ channel-blocking drugs (class I), β-blockers (class II), K+ channel blockers (class III) that prolong action potential (AP) duration (APD) and refractoriness, and L-type Ca2+ channel blockers (class IV). However, most AADs are “dirty drugs” that in a narrow range of concentrations simultaneously block different types of channels and modulate adrenergic and/or muscarinic receptors (e.g., quinidine, disopyramide, propafenone, amiodarone, dronedarone). Moreover, some old (adenosine, digoxin) and new AADs (ivabradine) cannot be listed in any of the original four classes. Table 54-2 shows drug selection based on the mechanisms underlying cardiac arrhythmias, and Figure 54-2 shows the main cellular targets for AADs. Table 54-1 Figure 54-2 Cellular targets of antiarrhythmic drugs (AADs). Most available AADs modify the conductance of Na+ (class I), Ca2+ (class IV), and K+ ion channels (class III) located in the sarcolemma, leading to a decrease or increase in a given ionic current, or they block β-adrenergic (class II), adenosine-A1, and muscarinic-M2 receptors. Some AADs inhibit abnormal persistent Na+ entry and diastolic Ca2+ release via the ryanodine receptor RyR2, with minimal effects on normal channel function. Furthermore, they can inhibit ion exchangers (Na+-Ca2+ [NCX]) or pumps (Na+-K+ ATPase), increase gap junction conductance, and correct ion channel expression within the membrane by modulating trafficking pathways. A1R, adenosine A1 receptor; AC, adenylyl cyclase; ATPase, Na+-K+ ATPase; β1AR, β1-adrenergic receptor; CaMKII, Ca2+/calmodulin kinase II; CSQ, calsequestrin; Gs, stimulatory G protein; M2R, muscarinic type-2 receptor; PBL, phospholamban; PKA, protein kinase A; RyR2, ryanodine receptor; SERCA2a, SR Ca2+-ATPase; SR, sarcoplasmic reticulum. Na+ channel blockers bind and unbind in a strongly time- and voltage-dependent manner to a receptor site within the pore-forming subunit (Nav1.5) of the channel when channels are in the activated or inactivated state.2 Na+ channel blockers also slow Na+ channel reactivation (transition from the inactivated to the resting state) upon repolarization, which prolongs refractoriness independently from changes in APD—that is, they increase the effective ratio of refractory period (ERP) to APD (postrepolarization refractoriness). As a consequence, the effects of class I AAD would be expected to increase: (1) at faster rates of stimulation (use-dependent block), because Na+ channels spend more time in activated and inactivated states, and the diastolic time for recovery from drug-induced block is shortened; and (2) in depolarized cardiac tissues (voltage-dependent block) because membrane depolarization inactivates Na+ channels and slows recovery from block. Clinical and experimental data suggest that block of atrial Na+ channels can terminate AF. Indeed, propafenone and flecainide are first-choice drugs for AF cardioversion, but only in patients without structural heart disease. Conversely, ventricular Na+ channel block is associated with proarrhythmic effects. Class I AADs decrease excitability and slow ventricular conduction and can increase all-cause mortality and, therefore, they have no role (or are contraindicated) in the primary prevention of sudden cardiac death (SCD) because of life-threatening ventricular tachyarrhythmias (VT or ventricular fibrillation [VF]) in high-risk patients with post–myocardial infarction.3 In secondary prevention, the use of class I AADs is limited because of their low efficacy and serious adverse effects that may offset the therapeutic benefit, including proarrhythmia, multiorgan toxicity, reduced cardiac function, or worsening coexisting diseases.4–6 Class I drugs are subdivided into drugs with intermediate (IA), fast (IB), and slow (IC) onset/offset kinetics of Na+ channel block (see Table 54-1). Recently, it has been demonstrated experimentally that drugs with fast onset/offset kinetics of Na+ channel block, such as vernakalant and ranolazine, could selectively target atrial versus ventricular Na+ channels.7,8 This targeting would allow them to exhibit a disease-specific component of atrial-selectivity because of the high atrial rate in AF, which enhances block of atrial over ventricular Na+ channels. Indeed, it has been proposed that the faster the AADs dissociation kinetics the more selective the atrial effect during AF and the fewer the ventricular proarrhythmic effects, because the fast dissociation kinetics allows recovery from block at slower frequencies (e.g., those of the ventricles either at sinus rhythm [SR] or during AF). Moreover, atrial cells exhibit a slightly more depolarized resting membrane potential (RMP) and a more gradual phase 3 of the AP, which at rapid atrial rates results in a less negative take-off potential.7,8 In addition, atrial cells exhibit a more negative potential for half-maximum inactivation of Na+ channels than ventricular myocytes. Because reactivation depends on membrane potential, fewer Na+ channels recover from the inactivated state during diastole in atria than in ventricles. Therefore, Na+ channel blockers that preferentially bind to the inactivated state of the channel and have a fast dissociation rate will exhibit atrial selectivity, because steady-state drug binding and consequently channel block will be larger in atria than in ventricles.8 However, block of K+ channels, particularly those preferentially or exclusively present in the atria, seems to be also required to meet the “class I atrial selective” profile. Otherwise lidocaine, which is highly selective for the inactivated state and exhibits a fast onset and offset kinetics, would be also atrial selective. Indeed, class IA and IC, like class III AADs, also block several K+ channels and prolong ventricular (quinidine) or atrial (propafenone and flecainide) APD, depending on the type of K+ channel they preferentially block (discussed later). Furthermore, amiodarone, dronedarone, ranolazine, and vernakalant, which produce an atrial-selective INa block and in animal models suppress AF with minor ventricular effects, also inhibit the rapid component of the delayed rectifier current (IKr) and other K+ currents prolonging atrial APD. This effect that enhances the use-dependent INa block by further reducing the diastolic interval at fast rates.8,9 Na+ channels open for a few milliseconds after membrane depolarization, generating peak INa, and then most of them undergo fast inactivation. However, some Na+ channels do not inactivate or inactivate but reopen during depolarization generating the late Na+ current (INaL), which presents slow inactivation and recovery kinetics.10 INaL increases under pathologic conditions, including myocardial ischemia, LV hypertrophy, HF, AF, and some variants of the long QT syndrome (LQTS).10,11 In the ventricular wall, M cells present a larger INaL than do epicardial and endocardial cells, and an increase in INaL increases transmural dispersion of repolarization (TDR) facilitating reentry.11 During myocardial ischemia, enhanced INaL increases Na+ influx and, via the Na+/Ca2+ exchanger (NCX), produces a Na+-mediated Ca2+ overload that prolongs ventricular APD. APD prolongation enhances triggered activity that can occur during phases 2 or 3 of the AP (early afterdepolarizations [EADs]), whereas intracellular Ca2+ (Cai2+) increase enhances triggered activity after AP repolarization (delayed afterdepolarizations [DADs]).10 Ranolazine is an antianginal drug that, besides its effects on peak INa at fast rates, blocks INaL and suppresses atrial and ventricular arrhythmias in a variety of conditions (e.g., ischemia, reperfusion, HF, AF, type 3 LQTS).10,11 During myocardial ischemia, ranolazine reduces intracellular Na+ and Ca2+ overload and improves mechanical dysfunction, but has no effect on INa, excitability or conduction velocity.11 In non−ST-elevation acute coronary syndromes12 ranolazine reduces non-sustained VT and supraventricular tachycardias, and preliminary evidence suggests that it safely converts paroxysmal AF and prevents AF recurrences.11 Moreover, ranolazine prevents ventricular APD (QT) prolongation, reduces TDR, and suppresses EADs produced by IKr inhibitors and in patients with type 3 LQTS. Describing the ultimate mechanisms responsible for the antiarrhythmic effects of class I AADs in reentry is a big challenge. Indeed, the underlying mechanism responsible for reentry itself is a matter of debate. Class I AADs markedly slow conduction and prolong refractoriness. The leading-circle theory of reentry predicts that because Na+ channel blockers slow conduction they should, if anything, favor reentry by decreasing the wavelength (product of refractory period and conduction velocity), resulting in proarrhythmic effects. The proarrhythmia risk increases with drugs that produce marked conduction slowing in depolarized-ischemic cardiac tissues. Experimental and clinical evidence, however, shows that class I AADs can terminate reentrant arrhythmias, such as AF, without increasing the wavelength. The most characteristic electrophysiological change that they produce before AF termination is an increase in the temporal excitable gap.13,14 According to the multiple wavelets theory of the origin of AF, this widening will lower the chance that reentrant waves encounter areas of partially refractory tissue, so that slowing of conduction and fractionation of wavelets will occur less frequently. This widening decreases the number of fibrillation waves by promoting their fusion, which increases the chance to terminate the arrhythmia. However, there are clinical and experimental data demonstrating that reentry is maintained by the periodic activity of one or a small number of functional reentrant sources with a wavefront rotating around a central core (“rotor”).15,16 The waves emerging from the rotors undergo spatially distributed fragmentation and give rise to fibrillatory conduction. The size of the spiral wave is determined by tissue excitability and refractoriness, so that the rotor will turn faster and in a more stable position, resulting in higher excitability and shorter refractoriness.15,16 In this context, Na+ channel blockers terminate reentry as they (1) enlarge center of rotation, so that the rotor cannot any longer be accommodated by the substrate; (2) decrease anchoring to functional obstacles, increasing meander and extinction at boundaries; and (3) reduce the number of daughter waves that could provide new primary rotors.15,16 β-Blockers include a heterogeneous group of drugs that inhibit sympathetic effects. Antiarrhythmic effects of β-blockers are the result of their electrophysiological effects (β-adrenoceptor stimulation modulates several ion currents, including hyperpolarization-activated inward current [If], INa, ICaL, IK1, Ito1, IKr, and IKs), inhibition of neurohumoral activation (and perhaps of sympathetic hyperinnervation and sprouting), and their antiischemic (reduce myocardial oxygen demands, increase subendocardial perfusion, and improve cardiac metabolism), antihypertensive, and antiproliferative effects.17,18 Indeed, β-blockers exert a beneficial effect on LV remodeling in patients with myocardial infarction (MI) or HF. Furthermore, in the ischemic myocardium, β-blockers decrease dispersion of ventricular repolarization and increase the VF threshold. Sympathetic hyperactivity is the predominant change in the autonomic tone preceding malignant ventricular arrhythmias and SCD, whereas reduced vagal activity is associated with increased mortality in HF. β-Blockers remain among the few AADs that are effective for both primary and secondary prevention of SCD in different clinical conditions, including acute and chronic myocardial ischemia, congestive HF or LV dysfunction, and hypertrophic cardiomyopathy.3,17,18 Thus, most patients who have a propensity to develop VT/VF should receive β-blockers because they are a rational, mechanism-based therapy for the treatment of these arrhythmias. Moreover, β-blockers decrease the incidence of postoperative AF, which is explained by the role of an increased adrenergic tone in the genesis of this arrhythmia. By inhibiting If and ICaL, β-blockers decrease spontaneous activity of the sinoatrial node (SAN), inhibit the ectopic activity of the His-Purkinje system (Purkinje fibers are particularly prone to develop abnormal automaticity during increased sympathetic activation), and suppress abnormal automaticity generated in cardiac cells depolarized to potentials between −60 and −40 mV owing to the activation of ICaL.18 In the atrioventricular node (AVN), they decrease conduction velocity and prolong refractoriness. β-Blockers are widely used to treat inappropriate sinus tachycardia and exercise-induced and supraventricular and ventricular arrhythmias where an increase in sympathetic tone plays a role17; they are the most effective drugs for ventricular rate control in AF and are first-choice drugs in the treatment of supraventricular reentrant tachycardias where the AVN forms part of the circuit.1,17 Finally, β-blockers are effective in controlling the proarrhythmia induced by class I AADs, probably because of their bradycardic effect, which decreases the rate-dependent conduction slowing induced by Na+ channel blockers. β-Blockers decrease all the proarrhythmic actions secondary to the increase of extracellular Ca2+ influx through L-type Ca2+ channels, sarcoplasmic reticulum Ca2+ release, and intracellular Ca2+ overload.17,18 Thus, they suppress EADs and DADs and are first-choice drugs in drug-induced torsades de pointes (TdP) and in patients with LQTS types 1 and 2 or catecholaminergic polymorphic VT (CPVT).19,20 Surprisingly, and even when there are marked pharmacologic differences among β-blockers, there are few direct comparator data regarding their efficacy. These drugs prolong cardiac APD and refractoriness effects that, under theoretical bases, can suppress reentry. Some drugs (e.g., dofetilide) are considered pure class III AADs because they selectively block the channels that generate the IKr (named Kv11.1 or HERG), thus prolonging both atrial and ventricular APD and refractoriness in the absence of effects in conduction velocity.9,21 Experimental data demonstrated that selective IKr inhibition produces a greater prolongation of APD and refractoriness in atria versus ventricles, at least at normal SR frequencies. Unfortunately, at fast driving frequencies, the relative role of IKr in atrial and ventricular repolarization diminishes so much that IKr inhibition is not able to prolong APD during the arrhythmia episodes. This property, known as reverse use-dependence, limits the clinical efficacy of these drugs to stop fast arrhythmias, particularly AF. Moreover, marked IKr inhibition produces excessive and inhomogeneous ventricular APD prolongation, results in TDR, and predisposes to EADs, which in turn can trigger TdP. Inhibition of IKr, and hence proarrhythmic risk, increases in the presence of bradycardia, hypokalemia, HF, or other IKr-inhibiting drugs. Indeed, many other drugs, including antihistamines, antipsychotics, antimicrobials, and diuretics, are able to markedly block Kr channels, thus prolonging the QT interval (drug-induced LQTS) and increasing the incidence of TdP and SCD. It has been proposed that prolongation of APD produced by pure IKr inhibitors is characterized by a set of four disturbances of repolarization including triangulation of the AP, reverse use-dependence, instability, and dispersion of APD whose magnitude would help to predict drug-induced proarrhythmia risk.22 HERG channel blockers gain access, from the intracellular side of the membrane, to a binding site located within the central cavity of the channel. The inner vestibule of HERG channels is larger than that of other K+ channels and presents two aromatic residues on each of the four subunits assembled to compose the conducting pore.23 These topological characteristics explain the marked pharmacologic promiscuity of HERG because they allow drugs with rather different chemical structures to establish interactions with the aromatic moieties by π electron stacking and block K+ efflux through the pore. Finally, it is worth mentioning that besides the “pure Kr blockers” many AADs, including amiodarone, quinidine, propafenone, flecainide, and ranolazine, also block HERG channels at therapeutic concentrations, an effect that could contribute to their antiarrhythmic properties particularly at the atrial level (i.e., amiodarone, ranolazine). However, unfortunately in some cases, HERG blockade can increase their ventricular proarrhythmic effects (quinidine). In healthy human atrial and ventricular myocytes driven at SR frequencies, IKs seems to be small, and its pharmacologic inhibition does not alter APD.24 Importantly, the role of IKs in determining APD rises in prominence at increasing beating frequencies or under β-adrenergic stimulation, which causes channel accumulation in closed states near the open state. In addition, IKs helps to terminate the AP when the repolarization reserve is compromised (i.e., IKr decrease).25 Moreover, recent data demonstrated that chronic AF increases by 100% IKs density in myocytes from both the right and left atria.26
Pharmacologic Bases of Antiarrhythmic Therapy
Antiarrhythmic Drugs: Introduction
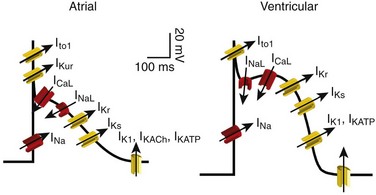
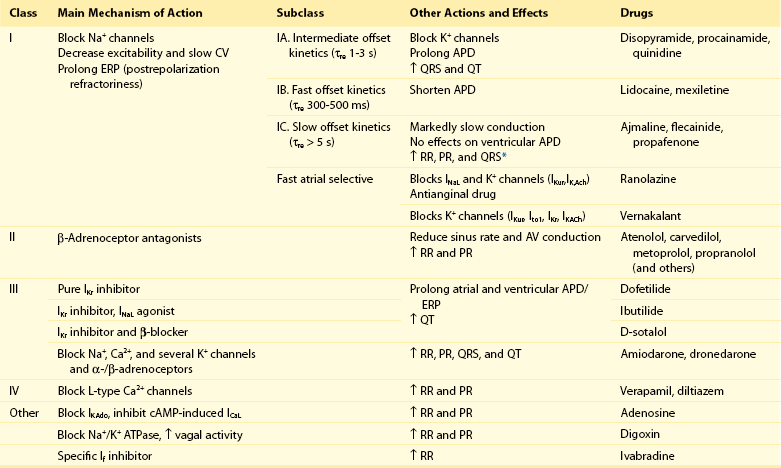
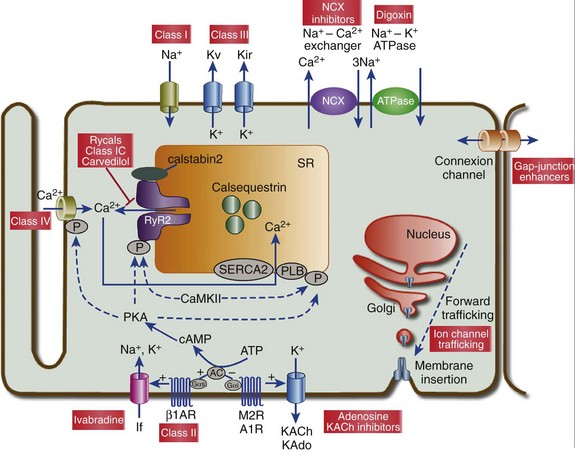
Na+ Channel Blockers: Class I AADs
Late Na+ Current (INaL) Inhibition
Antiarrhythmic Effects of Class I AADs
β-BLOCKERS: CLASS II AADs
K+ Channel Blockers: Class III Antiarrhythmic Effects
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Thoracic Key
Fastest Thoracic Insight Engine