Chapter 9 Pathophysiology of Vasculitis
The pathophysiological mechanisms that underlie vasculitis include elements of virtually all effector limbs of host defenses, including innate and adaptive immunity. Various classification schemes separate the vasculitides into primary and secondary families, and categorize them by size of the afflicted vessel. Classification of vasculitides is currently in ferment, with the classical American College of Rheumatology classification and the Chapel Hill Consensus Conference criteria under reconsideration from several fronts and for various reasons.1–3 Epidemiological studies and clinical trials, for example, require standardized definitions of these diseases, and these may overlap considerably. The confusion that reigns in this field reflects in part an incomplete understanding of the fundamentals of the pathogenesis of human vasculitides—an endeavor still in considerable flux. Whereas Chapter 41 provides a detailed classification of the vasculitides, this chapter focuses on the primary vasculitides and, for pedagogical purposes, considers the pathophysiological mechanisms of vasculitis in two broad categories: (1) mechanisms that underlie small-vessel vasculitis, and (2) mechanisms involved in medium- and large-sized arteritides (Fig. 9-1). Although this is an oversimplified categorization, it provides an organizational framework for discussing elements of humoral immunity involved chiefly in primary small-vessel vasculitides, and cellular immunity, likely the central mechanism underlying vasculitides of medium- and large-sized arteries (Table 9-1).
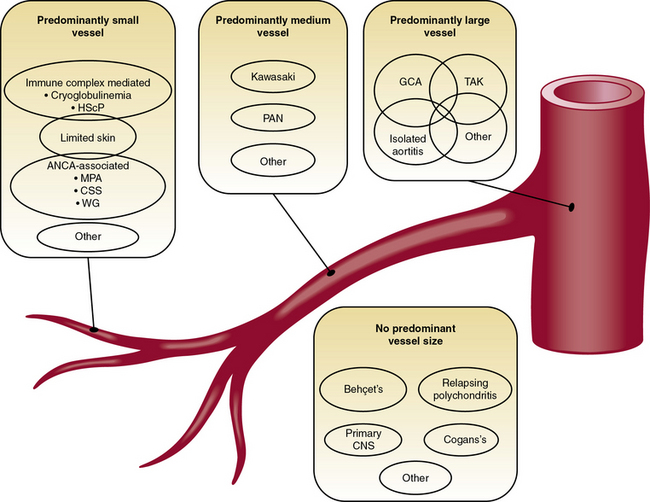
Figure 9-1 Classification scheme for the primary vasculitides.
(Modified from Suppiah R, Basu N, Watts RA, et al: Advances in the classification of primary systemic vasculitis. Int J Adv Rheumatol 7:10, 2009. Reproduced with permission from Watts RA, Suppiah R, Merkel PA, et al: Systemic vasculitis—Is it time to reclassify? Rheumatology 50:643, 2011.)
Table 9-1 Pathophysiological Mechanisms of Some Primary Vasculitides
| Putative Pathogenic Effectors | |
|---|---|
| Large-Artery Arteritides | |
| Takayasu arteritis | γδ T cells |
| Giant-cell arteritis | TH1 and TH17 helper T cells |
| Small-Artery Arteritides | |
| Microscopic polyangiitis | Anti-MPO ANCA > Anti-PR3 ANCA |
| Wegener granulomatosis | Anti-PR3 ANCA > Anti-MPO ANCA |
| Henoch-Schönlein purpura | Viral infections, food allergies (?) |
| Churg-Strauss’ syndrome | Eosinophils, CD95 |
| Cryoglobulinemia | IgM > IgG, hepatitis C virus infection |
ANCA, antineutrophil cytoplasmic antibody; Ig, immunoglobulin; MPO, myeloperoxidase; PR3, proteinase-3; TH, helper T cell.
Pathophysiology of Small-Vessel Vasculitis
Small-vessel vasculitides include Wegener granulomatosis, Churg-Strauss’ syndrome, microscopic polyangiitis, Henoch-Schönlein purpura, and essential cryoglobulinemic vasculitis.2,3 Recognition that antineutrophil cytoplasmic antibodies (ANCAs) associate with many (but not all) small-vessel vasculitides has advanced understanding of their pathophysiology. In particular, Wegener granulomatosis, microscopic polyangiitis, and Churg-Strauss’ syndrome associate strongly with ANCA. Many of the ANCA-positive small-vessel vasculitides involve the kidney.4
The principal antigens recognized by ANCA are the neutrophil enzymes myeloperoxidase (MPO) and proteinase-3 (PR3) (see Table 9-1); some ANCA may recognize human neutrophil elastase as well. Anti-PR3 antibodies also may recognize plasminogen.5 Antigens recognized by ANCA usually localize within polymorphonuclear (PMNs) leukocytes. When primed by stimuli such as tumor necrosis factor (TNF)-α or when undergoing apoptosis or NETosis (release of chromatin fibers called neutrophil extracellular traps (NETs) that trap and kill microbes extracellularly), PMN leukocytes can release these antigens. These antigens in turn can bind back to the cell surface—in the case of PR3, through CD1776—or decorate NETs.7 Binding of ANCAs to cell surface–associated MPO and PR3 on intact neutrophils leads to further activation of these leukocytes (i.e., generation of reactive oxygen species [ROS], release of lytic enzymes, binding of cells to the endothelium), as does engagement of the surface-bound Fc-portion of immunoglobulin (Ig)G in immune complexes with FcγRs on neighboring cells. Uptake of neutrophil-released MPO and PR3 by endothelial cells (ECs) also may impair the viability and vasomotor responses of ECs.8 These events together aggravate the local inflammatory response. As opposed to secondary vasculitides, which characteristically have substantial immune complex deposition upon histological examination, lesions of ANCA-associated conditions show modest Ig deposits.9 When released in soluble form, proteinases such as PR3 and neutrophil elastase readily bind to widely distributed and abundant antiproteinases that may mask their recognition by ANCA. Circulating ANCA can also complex with these antigens, but such complexes form preferentially when proteinase antigens remain associated with the neutrophil cell surface (Fig. 9-2).
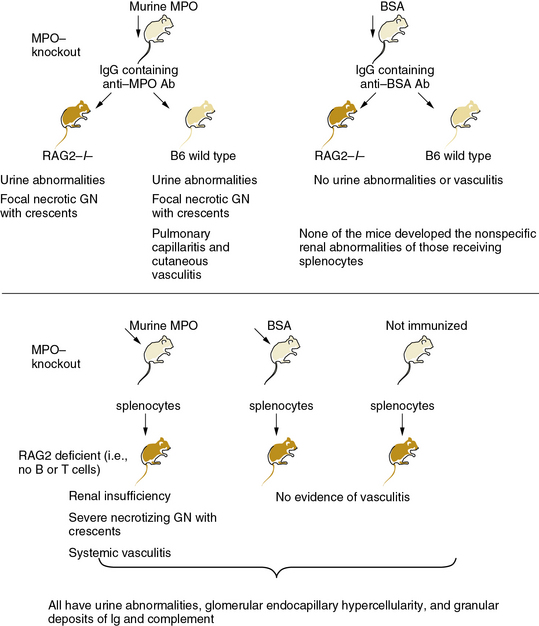
Figure 9-2 MPO-ANCA mice cause small-vessel arteritis.
(Data from Xiao H, Herringa P, Hu P, et al: Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest 110:955, 2002. Reproduced with permission from Day CJ, Hewins P, Savage CO: New directions in the pathogenesis of ANCA-associated vasculitis. Clin Exp Rheumatol 21:S35, 2003.)
Individuals who express primarily MPO-directed ANCA (vs. PR3-directed ANCA) may have distinct clinical courses.10 Microscopic polyangiitis associates particularly with anti-MPO ANCA, while Wegener granulomatosis typically associates with anti-PR3 ANCA (see Table 9-1). The possible clinical dichotomy between these patient categories may relate to the functions of target antigens. For example, binding to ANCA may protect MPO from clearance and inactivation by ceruloplasmin, increasing the ability of this enzyme to produce the highly oxidant species, hypochlorous acid (HOCl). Hypochlorous acid has many properties that may contribute to the pathophysiology of vasculitis, including stimulation of endothelial apoptosis.11
Not all patients with small-vessel vasculitis have ANCA-positive serology, indicating that some small-vessel vasculitides may involve other mechanisms or have low titer antibodies. Additionally, “atypical” ANCA directed against antigens other than MPO or PR3 may participate in the pathogenesis of vasculitis. Recent studies have implicated lysosomal-associated membrane protein-2 (LAMP-2) as a novel autoantigen in vasculitis.6,12,13 In addition to neutrophils, endothelial and other cells express LAMP-2, a recognition target for ANCA.
Antineutrophil cytoplasmic antibodies have proven unequivocally pathogenic in mice. In a landmark investigation, Xiao et al. immunized mice lacking endogenous MPO, owing to targeted gene inactivation, with exogenous mouse MPO.14 Transfer of splenocytes from these MPO-immunized mice into immunodeficient mice caused severe necrotizing crescentic glomerulonephritis. In some cases, a systemic necrotizing and granulomatous vasculitis affected lung capillaries as well as the renal microvasculature (see Fig. 9-2). Purified anti-MPO IgG isolated from the MPO-immunized mice caused renal, pulmonary, and cutaneous small-vessel vasculitis. Experimental depletion of neutrophils abrogated formation of glomerular lesions in anti-MPO IgG-treated mice, thus implicating granulocytes in the pathogenesis of ANCA-induced angiitis.15 Moreover, studies in bone marrow chimeric mice (transplantation of MPO wild-type or MPO-deficient bone marrow into MPO-immunized MPO-null animals) suggest that leukocytes are targets of anti-MPO ANCA.16
Immunization of PR3-deficient mice with PR3 yielded circulating anti-PR3 antibodies and modest renal and pulmonary vasculitis.17 These mice with anti-PR3 antibodies developed cutaneous vasculitis at sites of TNF-α injection. While not directly comparable to the passive/adoptive transfer studies in MPO-deficient mice, these results support different pathogenic capabilities of these two major classes of ANCA in terms of severity and localization of vasculitis, at least in mice. Plasma exchange causes improvement in patients with ANCA-associated disease exacerbation, supporting the causal role of antibody in these conditions.6
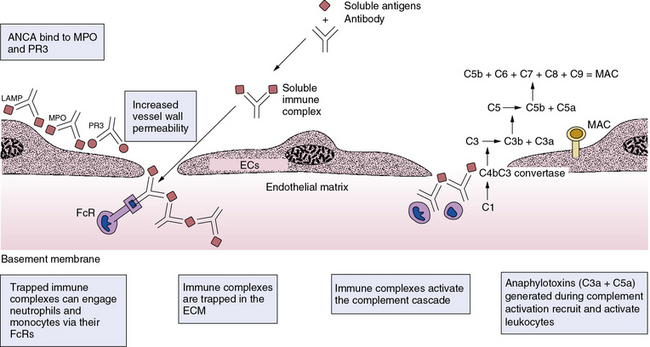
Antineutrophil cytoplasmic antibodies may provoke vasculitis in several ways (Figs. 9-3 and 9-4). These autoantibodies may increase activation and adherence of neutrophils to ECs.18 When neutrophils “primed” by exposure to TNF-α encounter MPO-ANCA, a respiratory burst can ensue and produce ROS such as superoxide anion and hydrogen peroxide—proinflammatory mediators that can injure ECs and activate smooth muscle cells (SMCs).19,20 Antineutrophil cytoplasmic antibodies promote neutrophil degranulation, and can activate intracellular signaling pathways and heighten sensitivity of PMN leukocytes to classic stimulants, such as formyl peptides.21 The mechanism of vascular damage in immune complex disease also involves complement activation (see Fig. 9-4).6 Antigen-antibody complexes (immune complexes) containing IgM or IgG can bind to complement factor 1 (C1), lead to assembly of C3 convertase, and yield activation of C3, C4, and C5. Ultimately, assembly of the membrane attack complex (MAC, composed of oligomers of C9 and other terminal complement components) can damage ECs by forming pores in their plasma membranes. Circulating immune complexes can sequester in subendothelial basement membranes at sites where interendothelial separation has occurred. These trapped immune complexes then activate complement, and can engage neutrophils and monocytes via their Fc and complement receptors. Anaphylatoxins (fragments of C3a, C4a, and C5a) generated during activation of the classical complement pathway can recruit granulocytes and monocytes and activate mast cells at sites of immune complex deposition in vessels. These leukocytes can amplify local inflammation and aggravate and perpetuate the vasculitic response.
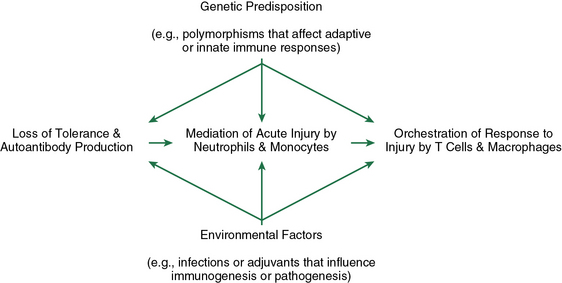
Figure 9-3 Pathogenic events during the evolution of ANCA disease.
(Reproduced with permission from Jennette JC, Xiao H, Falk RJ. Pathogenesis of vascular inflammation by anti-neutrophil cytoplasmic antibodies. J Am Soc Nephrol 17:1235, 2006.)
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


