Pathophysiology of Myasthenia Gravis
Jun Li
Robert P. Lisak
Myasthenia gravis (MG) is considered the prototypic autoimmune disease. Key features of the disease are muscle fatigability, decremental motor response to repetitive electrical stimulation, symptomatic improvement with drugs that inhibit acetylcholine esterase and the presence of antiacetylcholine receptor (AChR) autoantibodies in the majority of patients with generalized MG.
Historically, the first description of MG is controversial. A clear systematic description of the disease was published in 1900.4 Subsequent electrophysiologic studies by Harvey and Masland11 provided important diagnostic features of the disease. By 1960s, α-bungarotoxin (BuTx) was isolated from krait snakes and found to bind to AChR specifically.2,3 This toxin enabled a purification of AChR from the electric organ of Torpedo, a genus of electric rays.19 An autoimmune mechanism was proved by a serendipitous observation in 1973. Rabbits immunized with Torpedo AChR became paralyzed while experimenters were attempting to produce antibodies to study AChR. Anti-AChR antibodies were demonstrated to cause the blocking of neuromuscular junction transmission.21 The causal role of autoimmune mechanisms in human MG was firmly established when anti-AChR antibodies were demonstrated in the serum of MG patients and MG was reproduced in animals with passive transfer of IgG from human MG serum or with AChR-specific monoclonal antibodies.9,10,15,16,21,23,28 This chapter reviews the molecular structure and physiology of AChR, neuromuscular junction signal transmission, and the pathophysiologic mechanism of MG.
Neuromuscular Junction Transmission
Axons of the motor neuron communicate with muscle cells through a specialized structure, called the neuromuscular junction (Fig. 181-1). The junction consists of three parts: (a) the presynaptic terminal, an enlarged part of a distal axon, which synthesizes, stores, and releases neurotransmitters, such as acetylcholine (ACh); (b) the synaptic cleft, a microscopic space between the pre- and postsynaptic terminals; and (c) the postsynaptic terminal, also called the end plate, where the receptors for neurotransmitters (such as AChR) are located. During development, when the motor nerve fiber approaches the muscle cell, it induces a specialized indentation of muscle cell surface, the end plate. Upon the activation of presynaptic motor axons by propagated electrical activities, there is concurrent calcium influx to the presynaptic axon. These events activate coordinated intra-axonal molecular processes, which drive the neurotransmitter vesicles containing ACh to the presynaptic terminal membrane. Fusion of vesicles with the presynaptic terminal membrane takes place and the ACh is released in a quantal fashion. ACh diffuses rapidly across the synaptic cleft, binds to AchR, and directly opens the ion channel of AChR (Fig. 181-1). An individual ACh ion channel behaves in all-or-none pattern and produces a fixed-amplitude of current (2.7 pA) when it opens.25 A normal end plate current reflects the sum of 200,000 ACh-channel current and generates an end plate potential (EPP) of about 70 mV. The amplitudes of end plate current or potential are large, but the current is not regenerative, unlike the current through voltage-gated ion channels. The depolarizing current through a voltage-gated ion channel, such as a sodium channel, may cause additional sodium channels to open. Thus the current from many sodium channels may sum up, reach the threshold, and produce action potentials. In contrast, the Ach-induced EPP is unable to activate additional AChR ion channels. EPP must recruit and activate the sodium channels in the vicinity of the postsynapses, leading to the production of action potentials. Thus the number of available postsynaptic AChRs is critical for successful postsynaptic signal production and propagation.
To secure the neuromuscular transmission, a normal neuromuscular junction usually has an abundant reservoir of necessary biological mechanisms, including an excess of AChR as well as voltage-gated sodium ion channels. This is the so-called safety factor (SF). One may quantitatively define the safety factor by a formula: SF = EPP/(action potential threshold – membrane potential).13 In MG, the SF is impaired because of the reduction of postsynaptic AChR by the antibodies to AChR, diminishing the amplitude of EPP. Impairment of the SF may cause the failure of neuromuscular transmission. Excitation of postsynaptic muscle cell does not occur, leading to weakness.
Molecular Structure of AChR
AChR is a ligand-gated ion channel which, in mature innervated muscles, is composed of several homologous subunits (Fig. 181-2A), including α2, β, δ, and ε. In fetal or denervated muscles,
the γ subunit replaces the ε subunit. Five subunits line up like a barrel around the ACh ion channel. The ACh binds at the junctions between α and ε or γ subunits and between α and δ subunits. Each subunit has four transmembrane domains: M1 through M4 (Fig. 181-2B). Variations of amino acid sequence between the different subunits are generally in the large cytoplasmic domains. Amino acid sequences for the transmembrane domains are generally conserved. Sequences of the M1 and M2 of each subunit form the lining of AChR ion channel. Amino acids from the α-subunit sequence 66 to 76 are the critical autogenic region for development of pathogenic AChR antibodies.24 Interestingly, antibodies bound to this region in humans generally do not impair the ACh binding to AChR; rather, they can fix complement and initiate the destruction of postsynaptic membranes. In addition, AChR bound to the antibodies may have an increased rate of internalization and degradation, resulting in reduced available AChR.1,14
the γ subunit replaces the ε subunit. Five subunits line up like a barrel around the ACh ion channel. The ACh binds at the junctions between α and ε or γ subunits and between α and δ subunits. Each subunit has four transmembrane domains: M1 through M4 (Fig. 181-2B). Variations of amino acid sequence between the different subunits are generally in the large cytoplasmic domains. Amino acid sequences for the transmembrane domains are generally conserved. Sequences of the M1 and M2 of each subunit form the lining of AChR ion channel. Amino acids from the α-subunit sequence 66 to 76 are the critical autogenic region for development of pathogenic AChR antibodies.24 Interestingly, antibodies bound to this region in humans generally do not impair the ACh binding to AChR; rather, they can fix complement and initiate the destruction of postsynaptic membranes. In addition, AChR bound to the antibodies may have an increased rate of internalization and degradation, resulting in reduced available AChR.1,14
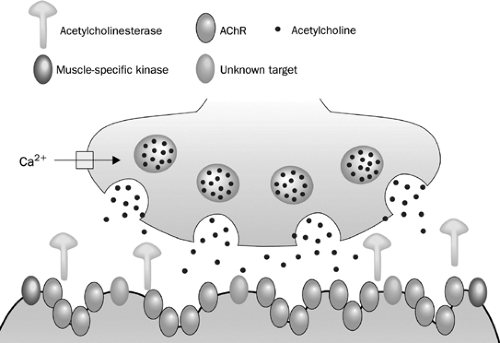 Figure 181-1. A presynaptic terminal is depicted on the top of the picture. ACh vesicles fuse with the terminal membrane after the presynaptic depolarization. The ACh is released, diffuses across the synaptic cleft and binds to the AChR. Once the AChR is activated, sodium influx occurs via the AChR ion channels, leading to the production of EPP. AChRs are the target of IgG antibodies in 85% of patients with seropositive MG; muscle-specific kinase (MuSK) antibodies are present in another 6%. The targets for other IgG or non-IgG plasma factors have yet to be identified. (From Vincent A et al. Lancet Neurology 2003;2:99–106, 2003.)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|