INTRODUCTION
Learning Objectives
The student will be able to define atelectasis and distinguish its various forms.
The student will be able to describe the pathophysiology and morphology of pulmonary edema, thromboembolism, hypertension, Goodpasture’s syndrome, and Wegener’s granulomatosis.
The student will be able to distinguish acute from chronic radiation pneumonitis and acute from chronic rejection of pulmonary allografts.
ATELECTASIS
Atelectasis describes a reduction in lung volume due to incomplete expansion of airspaces or, more commonly, the collapse of previously inflated pulmonary parenchyma. In atelectasis, perfusion of such nonventilated lung creates a physiological shunt that mixes inadequately oxygenated blood from pulmonary arteries with better oxygenated blood in the pulmonary veins (Chap. 9). If the ventilation-perfusion mismatch is sufficiently severe, systemic hypoxemia results. Additionally, atelectatic lung is more likely to become infected. Atelectasis can be subdivided by its pathogenesis.
In resorption atelectasis (also known as obstruction atelectasis), air is prevented from reaching distal airspaces because of airway obstruction. Then, as more distal air is absorbed, the previously expanded lung collapses. The extent of involvement is determined by the level of obstruction: Obstruction of a major airway can result in collapse of an entire lobe. Most commonly, a mucous or mucopurulent plug is responsible for the obstruction, although any physical obstruction will suffice. In resorption atelectasis, the mediastinum shifts toward the affected side.
In compression atelectasis (also known as passive atelectasis and as relaxation atelectasis), accumulation of space-occupying material within the pleural space mechanically compresses the lung parenchyma (Fig. 26.1). Compression atelectasis complicates pleural effusion and pneumothorax (see below), as well as pleural tumors (Chap. 31). Additionally, compression atelectasis of basal lung zones complicates peritoneal effusion (ascites) and frequently occurs in bedridden patients due to diaphragmatic elevation. In compression atelectasis, the mediastinum shifts away from the affected side.
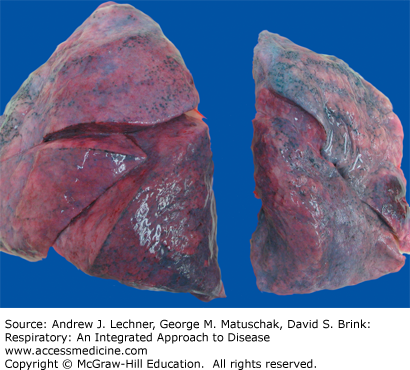
FIGURE 26.1
Compression atelectasis. The left lung (right side of image) is atelectatic due to a left-sided hemothorax caused by a gunshot wound. Note that the left lung is significantly smaller than the right and has a wrinkled pleural surface. From Kemp et al. Pathology: The Big Picture, McGraw-Hill; 2008.
Microatelectasis complicates adult and neonatal respiratory distress syndromes (Chaps. 23 and 39) as well as interstitial inflammatory lung diseases (Chap. 23). Its pathogenesis involves a complex set of events, the most important being deactivation of surfactant in the mature lung or its inadequate synthesis in neonatal lung.
In the setting of localized or generalized pulmonary fibrosis, the foci of fibrosis contract largely due to the action of myofibroblasts, collapsing adjacent lung tissue and resulting in contraction atelectasis or cicatrization atelectasis.
Generally, resorption atelectasis, compression atelectasis, and microatelectasis are reversible, while contraction atelectasis is not.
PULMONARY EDEMA
When edema develops in the lung, the excess fluid rapidly moves from the interstitium to the airspaces (Chaps. 7 and 28). While there are many specific causes of pulmonary edema (see Table 7.2), it is generally due to a change in hemodynamics (perfusion vs interstitial pressures) or to microvascular injury. Congestive heart failure is the most common cause of an increase in pulmonary venous pressure and the resultant pulmonary edema. Less commonly, reduction of plasma oncotic pressure will result in egress of fluid from the vascular space into the interstitium and then the alveoli. Alternatively, an increase in the permeability of capillaries, as occurs in the setting of alveolar and microvascular injury, can result in edema (Chap. 28).
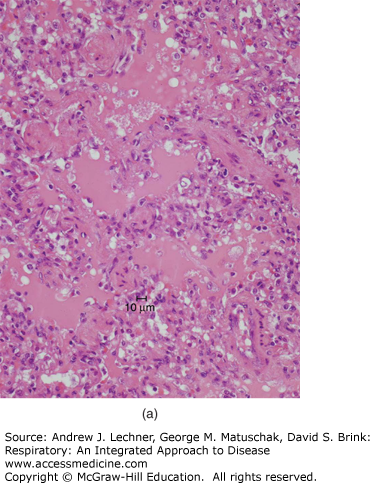
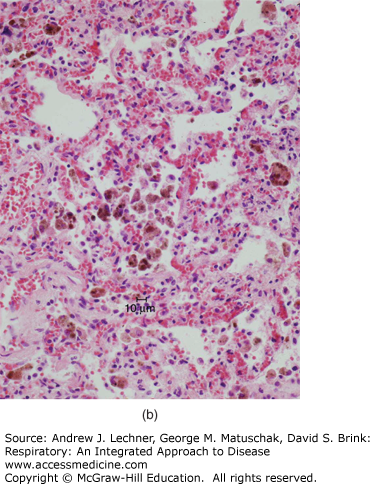
Gross manifestations of pulmonary edema include increase in lung weight and a wet cut surface with frothy fluid visible in larger airways. Microscopically, pulmonary edema is characterized by pale, eosinophilic, glassy or finely granular intra-alveolar precipitate [Fig. 26.2 (a)]. In the setting of pulmonary edema secondary to elevation of venous pressure, alveolar septal capillaries will show congestion characterized by engorgement with blood. Due to the delicate nature of alveolar septa, congestion is typically accompanied by occasional rupture of capillaries, resulting in microhemorrhages that release formed blood elements into alveolar spaces. As erythrocytes are cleared by macrophages, hemoglobin is progressively catabolized to hemosiderin, which persists in macrophage cytoplasm. Thus, the presence of hemosiderin-laden macrophages (or siderophages) reflects remote hemorrhage and implies chronic congestion [Fig. 26.2 (b)]. While any form of pulmonary hemorrhage will eventually show siderophages, the most common cause of pulmonary congestion and hemorrhage is congestive heart failure. This has led to widespread usage of the term “heart failure cells” to describe such intra-alveolar hemosiderin-laden macrophages.
FIGURE 26.2
(a) Pulmonary edema is recognizable microscopically as a pale, eosinophilic, glassy or finely granular precipitate filling airspaces. (b) In pulmonary congestion, alveolar capillaries are engorged with blood. Additionally, numerous hemosiderin-laden macrophages (“heart failure cells”) are present within alveoli, reflective of remote hemorrhage.
PULMONARY EMBOLUS
A large majority of pulmonary emboli are thrombotic in origin. Therefore, unless otherwise specified, the term “pulmonary embolus” typically refers to pulmonary thromboembolus. Pulmonary thromboembolism is involved in approximately 10% of hospital deaths. In more than 95% of cases, the source of pulmonary thromboemboli is a thrombus in a deep vein of the lower extremity (Chap. 27). Risk factors for venous thrombosis and pulmonary thromboembolus include prolonged bed rest (especially in the setting of lower extremity immobilization), severe trauma, burns, congestive heart failure, and hypercoagulable states (Table 26.1).
| Primary | Secondary |
|---|---|
| Antithrombin III deficiency | Obesity |
| Protein C deficiency | Recent surgery |
| Defective fibrinolysis | Pregnancy |
| Factor V Leiden | Oral contraceptive with high estrogen content |
| Prothrombin 20210A | Cancer |
| Hyperhomocysteinemia | |
| Antiphospholipid syndrome |
When a venous thrombus embolizes, it travels in progressively larger systemic veins toward the right heart. Upon ejection from the right ventricle, fragments of the embolism move into progressively smaller branches of the pulmonary artery until they reach vessels too small to allow passage. At that point the thromboemboli occlude a pulmonary artery or arteriole and increase pulmonary vascular resistance and can induce vasospasm. When a major vessel is occluded, the resulting pulmonary hypertension can reduce cardiac output and induce cor pulmonale and death. If death does not result, pulmonary thromboembolism results in hypoxemia due to ventilation-perfusion mismatch with increased dead space ventilation (Chap. 8). The ischemia may also reduce surfactant release and cause pleuritic pain that add to the work of breathing (Chaps. 5 and 6). Despite the lung’s systemic blood supply (Chap. 2), pulmonary thromboembolism can result in lung parenchymal ischemia and pulmonary infarction. In patients with patent foramen ovale (~30% of all people), pulmonary arterial thrombotic occlusion can cause right-to-left shunting and subsequent paradoxic embolism, in which a venous thrombus enters and embolizes systemic arteries and cause distal ischemia.
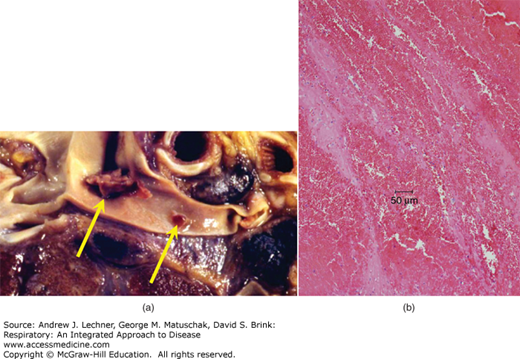
Grossly, a pulmonary thromboembolus is a serpentine blood clot impacted in a pulmonary arterial branch [Fig. 26.3 (a)]. A very large thromboembolus occluding the main pulmonary arteries is typically referred to as a “saddle embolus.” Microscopically, the thromboembolus is characterized by alternating layers of fibrin (eosinophilic) and erythrocytes. Ischemic lung damage is characterized by intra-alveolar hemorrhage [Fig. 26.3 (b)]. A pulmonary infarct will be conical (or, on cut section, wedge-shaped) and hemorrhagic [Fig. 26.4 (a)]. Initially, the infarct is red-blue. It becomes paler and later red-brown as erythrocytes lyse and hemoglobin is degraded to hemosiderin. Next, as fibroblasts progressively replace necrotic tissue with scar, the infarct shows a gray-white peripheral zone. Microscopically, infarcted lung tissue shows coagulative necrosis with loss of nuclear basophilia, although alveolar hemorrhage often dominates the microscopic appearance [Fig. 26.4 (b), (c)]. In cases of sudden death due to saddle embolus, there are typically no gross or microscopic changes to the lung.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


