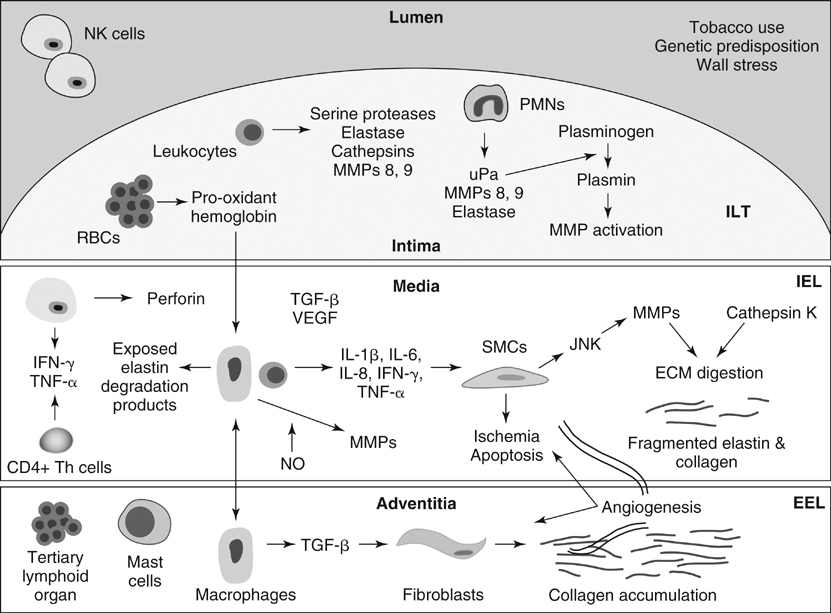
MMPs play an integral role in the inflammatory processes associated with AAA formation ( Figure 1 ). The MMPs include at least 15 structurally related proteinases that are a subfamily of the metazincin superfamily of proteinases. Substrates of the MMPs include elastin, noncollagenase extracellular matrix (ECM) proteins, including fibronectin and laminin, and nonstructural components. The nonstructural components include interleukin (IL)-1α, IL-1β, IL-2 receptor, active MMP9, chemokine ligand 5, TGF-β, and several of the pro-MMPs. Four MMPs exhibit specific activity with elastin, including MMPs 2, 7, 9, and 12. It has been proposed that as the ECM is digested, signals in the form of cryptic fragments may be released, including arrestin, deprellin, endostatin, restin, elastin degradation products, and growth factors TGF-β and vascular endothelial growth factor (VEGF).
FIGURE 1 Schematic representation of abdominal aortic aneurysm development. ECM , Extracellular matrix; EEL , external elastic lamina; VEGF , vascular endothelial growth factor; IEL , internal elastic lamina; IFN- γ, interferon-γ; IL , interleukin; ILT , intraluminal thrombus; JNK , c-Jun N-terminal kinase; MMP , matrix metalloproteinase; NK , natural killer cell; NO , nitric oxide; PMN , polymorphonuclear cell; RBC , red blood cell; SMC , smooth muscle cell; TGF- β, transforming growth factor-β; Th , T-helper lymphocyte; TNF- α, tumor necrosis factor-α; uPa , urokinase-type plasminogen activator. Numerous MMPS including 1, 2, 3, 8, 9, 10, 12, and 13 play roles in AAA formation. MMPs 1, 8, and 13 are capable of initiating the degradation of fibrillar collagen, and MMPs 2 and 9, both overexpressed in human and experimental AAAs, have both elastolytic and collagenolytic properties. The presence of constitutive MMP2 in smaller AAAs suggests a possible role for MMP2 in early aneurysm formation. MMP9 is not typically produced in normal aorta, but it is present in atherosclerotic plaques, suggesting a possible role in plaque rupture. MMP9 expression is increased in the serum and aortic tissue of AAA patients compared to that of patients with aortoiliac occlusive disease. There is a correlation between MMP9 expression and AAA size, and it appears to play a role in AAA expansion and ultimate rupture. Furthermore, endovascular exclusion of AAAs results in decreased MMP9 levels. The expression of MMP12 is also increased in AAA tissue, and an association with aortic media elastin has been identified.
In terms of experimental animal models, MMP9 knockout mice do not form AAAs; however, after undergoing wild-type bone marrow transplantation, the same MMP9 knockout mice exhibit an aneurysm phenotype. Secreted tissue inhibitors of metalloproteinases (TIMPs) 1, 2, and 3, as well as plasma-derived α-1-macroglobulin, cause direct inhibition of MMPs. Inflammatory states associated with increased MMP expression typically result in increased TIMP1 levels. Inhibition of the MMPs with exogenous TIMPs and α-2-macroglobulin have attenuated the development of AAAs in animal models. It has been demonstrated that doxycycline, a tetracycline antibiotic, is a nonselective MMP inhibitor, and nonantibiotic tetracycline derivatives have demonstrated MMP inhibition and have been used to prevent AAA formation in animal models and in small human studies.
The cystine proteases, including cathepsins K, L, and S, also play roles in AAA formation. The expression of all cathepsins is increased in human AAA tissue compared to normal aortic tissue. Cathepsin K is the most potent elastolytic enzyme. Cystatin C plays the greatest inhibitory role against the cathepsins, and it is ubiquitously expressed throughout the human body. The constitutive expression of cystatin C is significantly decreased in AAA patients. Furthermore, immunohistochemical analyses of human AAA tissue have demonstrated increases in cathepsin S expression and decreases in cystatin C expression compared to that in normal AAA tissue. Cathepsin S–deficient mice demonstrate less aortic dilation after elastase exposure compared to wild-type mice.