Management and Therapeutic Interventions |
INTRODUCTION
Oxygen was discovered in the late 18th century simultaneously by several investigators. Joseph Priestley, a minister in England, made his discovery after attempting to melt mercury oxide using a magnifying glass and the sun’s rays. The vapor that was produced by this melting allowed a candle to burn brighter, and Priestley later discovered the vapor could be used in place of air to keep a mouse alive. He first published his findings in 1774. At about the same time, and in a similar way (i.e., by burning mercury oxide), Carl Scheele, a pharmacist in Sweden, described a gas he called “fire air.” He may have done his experiments before Priestley, but he did not publish his findings until 1777. Finally, after being told about this new vapor during a 1774 visit by Joseph Priestley, the chemist, Antoine Lavoisier, devised quantitative experiments which he used to prove the existence of this element and its role in combustion. He named the element “oxygen” and published his research in 1777.
Soon after its discovery, oxygen was being utilized as a medicine and cure-all for many respiratory diseases, including “consumption” and asthma. However, suspicions about the safety of oxygen therapy were raised almost as soon as it was discovered and even before the element was named. In 1775, Joseph Priestley wrote of oxygen (which, at that time, he called “dephlogisticated air”): “A moralist, at least, may say that the air which nature has provided for us is as good as we deserve.”1
Rigorous scientific evaluation of the potential toxicities of oxygen did not occur until the work of James Lorrain Smith, an Edinburgh pathologist, who first published the pulmonary pathologic alterations associated with oxygen exposure in 1899. It was also recognized around this time that under ambient air, the arterial blood was already at near-maximal oxygen-carrying capacity, putting into doubt the utility of further increasing the fraction of inspired oxygen.
Following these observations, the use of therapeutic oxygen fell into some disrepute.
In the early 1920s, supplemental oxygen therapy was reevaluated yet again. This was occurring as the detrimental effects of tissue hypoxia were being recognized, along with their reversibility with supplemental oxygen. Since the mid-20th century, especially with the advent of improved oxygen delivery systems, mechanical ventilation, and the modern intensive care unit, the use of oxygen has become a standard prescribed therapy for a multitude of cardiac and respiratory diseases. Oxygen therapy is now common in the outpatient setting. As of 2005 in the United States, approximately 1 million people receive long-term oxygen therapy (LTOT) from the Medicare Program, with total reimbursement costs related to LTOT exceeding 2 billion dollars per year.2
In this chapter, the physiology of hypoxia at the tissue level and mechanisms of arterial hypoxemia are discussed. In addition, various clinical assessments of hypoxemia, indications for acute and LTOT, and available methods of noninvasive oxygen administration are considered. Finally, the pathophysiology and clinical manifestations of pulmonary oxygen toxicity are addressed, along with the potential for oxygen toxicity in other organ systems.
TISSUE OXYGENATION
Important to remember is that the primary goal of supplemental oxygen therapy is to ensure appropriate oxygen delivery to vital end-organ tissues. Therefore, one must understand the mechanisms of tissue hypoxia to understand the indications for using supplemental oxygen.
Tissue hypoxia is governed by the balance between oxygen delivery and oxygen utilization. Generally, failures in oxygen delivery lead to most instances of tissue hypoxia. Oxygen delivery is determined by the cardiac output and the oxygen content of arterial blood, as described by the formula:
![]()
where CaO2 is calculated by the formula:
![]()
where SaO2 is the arterial hemoglobin oxygen saturation and PaO2 is the oxygen tension or partial pressure of arterial blood.
From this formula, the causes of poor oxygen delivery can be narrowed to three categories: Low cardiac output states (i.e., various forms of shock), low hemoglobin concentration states (i.e., anemia), and low SaO2 states (i.e., arterial hypoxemia or hemoglobinopathies). Note that a low oxygen tension, although often leading to low arterial hemoglobin saturation, does not contribute appreciably to oxygen delivery. This can be seen numerically in the above formula for CaO2, where the PaO2 is multiplied by 0.0031 and, therefore, contributes little to the final product. It is important to point out that supplemental oxygen therapy addresses only arterial hypoxemia. For other causes of tissue hypoxia, more specific therapies are generally required, such as vasoactive agents or intravenous fluid administration to reverse shock, and blood transfusions for anemia.
Along with the primary basis of poor oxygen delivery, tissue hypoxia can also be caused by inappropriate or excessive utilization of oxygen. One cause of inappropriate oxygen utilization is histotoxic hypoxia, which occurs when cells, for a variety of reasons, are unable to utilize oxygen for aerobic metabolic respiration. This is classically seen in cyanide poisoning, where the cyanide molecule inhibits the enzyme cytochrome c oxidase, and the cell is subsequently unable to use oxygen to produce adenosine triphosphate (ATP) via the mitochondrial electron transport chain. This leads to glycolysis, profound lactic acidosis, and cell and tissue death. Administration of oxygen is indicated in this condition, but it will not, by itself, reverse the toxicity of cyanide; other antidotes must be given (generally, sodium nitrite, amyl nitrite, and sodium thiosulfate).3
Another cause of decreased oxygen utilization may occur in the setting of sepsis. In this case, oxygen is not utilized by tissues due to both mitochondrial damage and abnormal shunting of blood from a variety of insults at the endothelial and microcirculatory levels. Finally, oxygen may be excessively utilized by tissues relative to oxygen delivery. This may occur in hypermetabolic states, such as thyrotoxicosis4 and fever.5
ARTERIAL HYPOXEMIA
Important is the distinction between the terms hypoxia and hypoxemia. Hypoxia may be thought of as the functional equivalent of ischemia and refers to a lack or misuse of oxygen at the tissue level. Hypoxemia refers to the blood’s oxygen content and can be more formally defined as a validated deficiency of oxygen tension in the blood. In other words, when a patient’s PaO2 and, therefore, SaO2 are low, the patient is hypoxemic. Since arterial hypoxemia is a major cause of tissue hypoxia, the cause of which is most likely to be reversed or improved with oxygen therapy, its major etiologies are discussed below.
The driving force of oxygen transport across the alveolar barrier into the blood depends on both the partial pressure of oxygen in the alveolus (PAO2) and overall respiratory function. Thus, arterial hypoxemia results from either reduction of the inspired oxygen tension or dysfunction of the respiratory system. Along with the reduction of inspired oxygen tension (such as at altitude), there are four other pathophysiologic causes of arterial hypoxemia (Table 144-1).
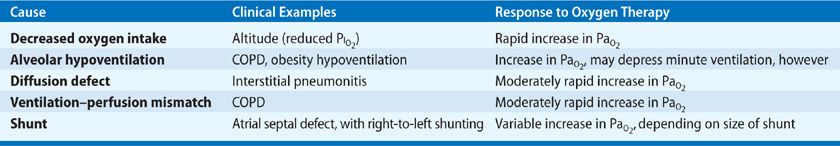
Alveolar hypoventilation leads to a diminished PAO2 and resultant decrease in PaO2 and SaO2. It may constitute the final common pathophysiologic denominator for a variety of pulmonary and nonpulmonary disorders leading to hypercapnia and hypoxemia. Examples of nonpulmonary disorders leading to these derangements include narcotic overdose, which reduces the central drive to breath; abdominal compartment syndrome, which can mechanically reduce ventilation due to abdominal distension; and obstructive sleep apnea, which is the result of transient loss of the upper airway patency.
Another pathophysiologic cause of hypoxemia arises with defects in diffusion of oxygen across the alveolar–capillary membrane, as, for example, in fibrotic lung diseases and interstitial pneumonias. The increased barrier to oxygen diffusion is detected as a reduced diffusion capacity of carbon monoxide (DLCO). Diffusion limitation generally coexists with ventilation–perfusion mismatch. Its effects on oxygenation often become clinically prominent in states of increased cardiac output (such as exercise) in the setting of fibrotic or interstitial lung disease.6 These higher output states lead to reduced time for diffusion of oxygen across the diseased alveolar–capillary membrane and subsequent hypoxemia.
Clinically, the most frequently observed pathophysiologic mechanisms of hypoxemia are ventilation–perfusion mismatch and shunt.
Ventilation–perfusion mismatch generally results when focal (or sometimes diffuse) areas of diminished ventilation arise relative to persistent pulmonary capillary blood flow. Clinical examples include cardiogenic pulmonary edema, acute lung injury, and pneumonia—all of which are characterized by alveolar filling, reduced ventilation, and continued perfusion. The hypoxemia resulting from lack of ventilation to these areas can generally be overcome simply by increasing the fraction of inspired oxygen (FIO2), that is, with the use of supplemental oxygen. In the setting of ventilation–perfusion mismatch, without oxygen supplementation, reflex hypoxic pulmonary vasoconstriction occurs, minimizing the impact on PaO2. Hypoxic pulmonary vasoconstriction may ultimately lead to increased pulmonary artery pressure and cor pulmonale.
Shunt occurs when a fraction of pulmonary blood flow effectively bypasses the alveolar compartment. Examples include atelectatic lung in which ventilation is absent but perfusion maintained (at least in part). Other examples include atrial septal defect and pulmonary arteriovenous malformations. Increasing the alveolar concentration of oxygen in the setting of shunt has no effect on arterial oxygenation. The hypoxemia from shunt physiology is refractory to the use of supplemental oxygen.
Finally, when tissue hypoxia is severe, or when increased tissue oxygen utilization is present, the oxygen content of venous blood returning to the right heart may be profoundly reduced—detected as a low oxygen saturation of venous blood (SVO2). A low SVO2, in combination with ventilation–perfusion mismatch or shunt, leads to worsening arterial hypoxemia. This pathophysiologic constellation may be observed when patients with severe lung disease and poor oxygen delivery are physically active or have high metabolic demands due to critical illness. Hemoglobin desaturation can then be seen.
CLINICAL ASSESSMENT OF TISSUE HYPOXIA
To use oxygen therapy correctly, clinicians must recognize the presence of tissue hypoxia and its etiology. With this knowledge, the clinician can determine who may benefit from supplemental oxygen. Furthermore, the degree of hypoxia must be assessed to determine the optimal dosage of oxygen. In this way, one can balance oxygen’s benefits and potential toxicities. Unfortunately, tissue hypoxia is not directly measured by any one test. Instead, a combination of clinical and laboratory parameters is used to determine its severity. In the case of tissue hypoxia caused by isolated arterial hypoxemia, measurement of arterial oxygen saturation can be an excellent guide to appropriate oxygen therapy.
 CLINICAL MANIFESTATIONS
CLINICAL MANIFESTATIONS
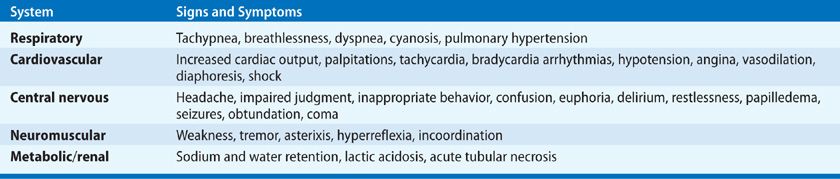
Clinical manifestations of hypoxia are variable and nonspecific. Whether hypoxia develops acutely or is chronic influences patient presentation: Patients with chronic hypoxia often have minimal symptoms. Symptoms and signs of acute hypoxia are outlined in (Table 144-2). The most prominent findings are dyspnea, tachypnea, and respiratory distress. A frequently observed finding is an acute change in mental status, ranging from agitation to delirium to coma. Cardiac arrhythmias are common. Cyanosis is generally a late finding and is often unreliable in diagnosing hypoxia, as it may be absent in the setting of anemia and with poor peripheral perfusion.
 LABORATORY ASSESSMENT
LABORATORY ASSESSMENT
Distinction between tissue hypoxia and arterial hypoxemia is important in applying various laboratory studies in clinical evaluation. To determine if tissue hypoxia, that is, hypoxia in end-organs, is present, measures such as serum lactate (elevated in ischemia) and SVO2 (reduced in ischemia) are generally used. Evaluation for anemia by measuring hemoglobin or hematocrit is also important in this context.
When the cause of tissue hypoxia is isolated arterial hypoxemia, measurements of PaO2 and SaO2 provide appropriate surrogates of oxygen delivery and, therefore, tissue hypoxia. Values are derived from arterial blood-gas measurements. Since SaO2 and PaO2 have a generally predictable relationship according to the oxygen–hemoglobin dissociation curve, measuring only SaO2 may be sufficient to assess arterial hypoxemia. This is done noninvasively using pulse oximetry; the estimated SaO2 is designated as SpO2. Importantly, various toxins, such as carbon monoxide and those that result in methemoglobinemia will disrupt the predictable relationship between SaO2 and PaO2 as measured by pulse oximetry (see next section). Under these circumstances, cooximetry performed on an arterial blood-gas sample is required, and pulse oximetry cannot be relied upon to evaluate arterial hypoxemia.
 PULSE OXIMETRY
PULSE OXIMETRY
Pulse oximetry is an important diagnostic tool which is now widely utilized in hospitals, outpatient settings, and at home. The general principles of this technology are useful to understand, as are its limitations.
First proposed conceptually in the 1930s and developed further in the mid-20th century, the underlying principle of oximetry is Beer’s law, which states that concentration of an unknown substance dissolved in a solvent can be determined by its light absorption7:
![]()
where

When light is passed through a finger, ear lobe, or other tissue, the majority of it is absorbed by connective tissue, skin, bone, and venous blood, and the amount absorbed is constant. However, with each heartbeat, pulsatile flow of arterial blood occurs, allowing the pulse oximeter to detect changes in light absorbance at two wavelengths, 660 nm (red) and 940 nm (infrared). Since reduced hemoglobin and oxyhemoglobin have different absorptions at these two wavelengths, the concentration of oxyhemoglobin relative to total hemoglobin can be deduced.
Several limitations to pulse oximetry exist, which can be inferred from its basic principles. Any substance in the blood with similar absorption at the two wavelengths employed will interfere with accuracy. This is clinically important in the evaluation of two acquired hemoglobinopathies—carboxyhemoglobinemia and methemoglobinemia.
Carboxyhemoglobin has a very similar absorption coefficient at 660 nm as oxyhemoglobin; at 940 nm, it has minimal light absorption. Therefore, no matter what the relative concentrations of hemoglobin molecules saturated with CO versus O2, SpO2 will be constant (generally about 90%–95%).
In methemoglobinemia, a condition in which the iron moiety of hemoglobin is reduced to the ferric state, the methemoglobin molecule is characterized by equalization of absorption coefficients at 660 nm and 940 nm. Equal absorption at these two wavelengths translates into an SaO2 of 85%, which is what the pulse oximeter inevitably reads as methemoglobin concentration increases. Furthermore, an antidote for methemoglobinemia, methylene blue, also affects absorption of light at 660 nm and may falsely lower SpO2.
Given that pulse oximetry relies upon pulsatile flow, the technology cannot be used in conditions of low flow, such as cardiac arrest or profound shock. Errors also occur when venous pulsatility is prominent, such as in severe tricuspid regurgitation. In this case, the pulsatility of venous blood, with its higher concentration of reduced hemoglobin, may result in artificially low readings of oxygen saturation. When profound arterial hypoxemia exists, for example, SaO2 <70%, the accuracy of pulse oximetry declines, as the technology has generally not been calibrated at lower saturations.
 OTHER ASSESSMENTS OF HYPOXEMIA
OTHER ASSESSMENTS OF HYPOXEMIA
Several other calculated assessments of hypoxemia are useful in assessing the degree of respiratory dysfunction.
PaO2/FIO2
PaO2/FIO2 (P/F ratio) requires an arterial blood-gas specimen. For example, consider a patient breathing gas with an FIO2 of 0.80, who has a PaO2 240 mm Hg. The P/F ratio is 240/0.8 or 300. The ratio, considered to reflect the magnitude of derangement in O2 transfer across the lung and thought to be relatively unaffected by differences in FIO2 among measurements, is most useful in mechanical ventilation, when accurate determination of FIO2 is possible. In the setting of acute hypoxemia, a P/F ratio of <300 indicates abnormal gas exchange, and a P/F ratio <200 indicates severe hypoxemia.
Alveolar–Arterial (A-a) Difference
The A-a difference (also called the A-a gradient) measures the difference in alveolar oxygen tension and arterial oxygen tension. This measurement, which requires an arterial blood gas, is determined as:
![]()
PAO2 is calculated using the alveolar air equation:
![]()
where,

A “normal” A-a difference on room air is often considered to be 10 mm Hg. However, two important factors affect the “normal” value. The first is patient’s age, the influence of which can be expressed numerically according to the formula:
![]()
The second is FIO2: The higher the FIO2, the larger the A-a difference, since alveolar oxygen tension increases more than does arterial oxygen tension with increasing FIO2. Note the distinction in this regard with respect to the P/F ratio described previously.
Oxygenation Index
The oxygenation index (OI) is often used in neonates and children with acute hypoxemic respiratory failure to assess the severity of disease and predict outcome.8 It can be calculated only for mechanically ventilated patients, where mean airway pressures may be measured.
![]()
A high OI (e.g., >25) indicates severe hypoxemic respiratory failure.
INDICATIONS FOR OXYGEN THERAPY
Supplemental oxygen is a drug with indications, contraindications, and a therapeutic window. Oxygen dosage should be titrated as precisely as possible and oxygen administration monitored closely with respect to clinical benefits and potential toxicities. Indications for oxygen therapy may be considered in both acute and chronic settings. In addition, special circumstances under which oxygen is used therapeutically warrant special discussion, including hyperbaric administration (oxygen delivered at >1 atmosphere) and oxygen therapy during air travel.
 ACUTE INDICATIONS FOR OXYGEN THERAPY
ACUTE INDICATIONS FOR OXYGEN THERAPY
Indications for acute, short-term supplemental oxygen therapy, listed in Table 144-3, are based upon guidelines established by the American Association of Respiratory Care and other organizations.9–11
Source: Data from Kallstrom TJ; American Association for Respiratory Care (AARC). AARC Clinical Practice Guideline: oxygen therapy for adults in the acute care facility–2002 revision & update. Respir Care. 2002;47(6):717–720; O’Driscoll BR, Howard LS, Davison AG; British Thoracic Society. BTS guideline for emergency oxygen use in adult patients. Thorax. 2008;63 Suppl 6:vi1–vi68; Fulmer JD, Snider GL. American College of Chest Physicians (ACCP)–National Heart, Lung, and Blood Institute (NHLBI) Conference on oxygen therapy. Arch Intern Med. 1984;144(8):1645–1655.
Usually, when a patient presents with respiratory distress, supplemental oxygen is administered until an assessment of hypoxemia is made. The clearest indication for oxygen therapy is arterial hypoxemia, and supplemental oxygen is generally administered when the PaO2 is <60 mm Hg, which normally corresponds to an SaO2 or SpO2 of 89% to 90%. When the PaO2 drops below 60 mm Hg, oxygen saturation may drop precipitously, leading to a much lower arterial oxygen content and resultant tissue hypoxia.
As noted previously, the most common pathophysiologic cause of hypoxemia is ventilation–perfusion mismatch, which responds readily to supplemental oxygen. Hypoxemia secondary to right-to-left shunt is less responsive to supplemental oxygen; when the shunt fraction exceeds 20% to 25%, hypoxemia persists, despite administration of inspired gas with an FIO2 of 100%. In the case of hypoxemia due to alveolar hypoventilation, oxygen therapy often improves SaO2 and oxygen delivery, but it does not correct the underlying cause of respiratory dysfunction; restoration of ventilation must be pursued.
An oxygen saturation target of 94% to 98% is reasonable for patients at sea level who are hypoxemic and who have normal baseline lung function.10 In certain circumstances, such as shock states characterized by hypoperfusion, the target may be even higher in an attempt to maximize the oxygen content of arterial blood. Conversely, in patients who have abnormal control of respiration, such as those with chronic hypercapnia, a lower PaO2 goal may be more appropriate (as discussed later in this chapter).
Use of supplemental oxygen may be indicated in a few clinical scenarios other than arterial hypoxemia. As discussed earlier, patients in shock states may benefit from enhanced arterial oxygen content and delivery, as may patients in cardiac and respiratory arrest. Similarly, trauma victims and critically ill surgical patients benefit from early oxygen therapy to improve tissue hypoxia. Use of short-term, postoperative supplemental oxygen has also been shown to reduce wound infection in surgical populations.12 In patients with carbon monoxide poisoning, in whom pulse oximetry readings are unreliable, supplemental oxygen increases PaO2. More importantly, it shortens the half-life of carboxyhemoglobin, thereby increasing the percentage of oxyhemoglobin. With administration of pure oxygen, the half-life of carboxyhemoglobin is 70 to 80 minutes,13 compared with a half-life of 320 minutes when a patient is breathing room air.14 Under hyperbaric conditions, when pure oxygen is delivered at higher atmospheric pressure, the half-life is shortened even further. Hyperbaric oxygen is generally employed when carboxyhemoglobin levels are high (>25%) and there is an evidence of end-organ damage, such as cardiac ischemia or alterations in sensorium,15 although use of hyperbaric oxygen in this setting is currently controversial.
Several other disorders may benefit from administration of supplemental oxygen, although supportive data in these settings are lacking or equivocal. Oxygen therapy is often used in cluster headaches,16 sickle cell pain crises (although this is falling out of favor),17 palliative relief of dyspnea without hypoxemia,18 and pneumothorax in patients who do not have a chest tube (in an effort to facilitate resorption of pleural air).19,20 Administration of supplemental oxygen has also been proposed to reduce postoperative nausea and vomiting and to alleviate nausea associated with medical transport in ambulances.21 Use of supplemental oxygen in uncomplicated myocardial infarction is common but controversial; the topic is discussed in more detail below. Finally, hyperbaric oxygen is sometimes used in an effort to accelerate healing of complicated, chronic wounds arising in the setting of peripheral or microvascular disease.22 Hyperbaric oxygen therapy is widely accepted as the therapy of choice in decompression sickness (see Chapter 93).23
 INDICATIONS FOR LONG-TERM OXYGEN THERAPY
INDICATIONS FOR LONG-TERM OXYGEN THERAPY
The use of LTOT has been growing as our ability to deliver oxygen in outpatient settings has improved. Criteria for administering LTOT have become well established, generally in the context of chronic obstructive pulmonary disease (COPD). Table 144-4 outlines the guidelines of the American Thoracic Society (ATS) for LTOT in COPD.24 The guidelines are based on the data derived from two major trials.
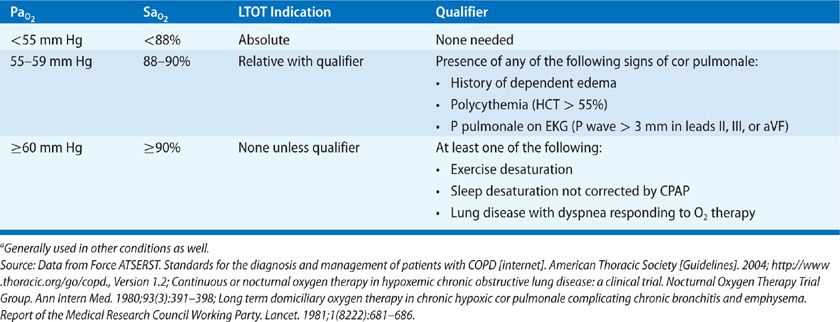
The first trial, published in 1980, is the Nocturnal Oxygen Therapy Trial (NOTT).25 In NOTT, 203 hypoxemic patients with COPD were randomized to the treatment with either nocturnal (at least 12 hours) or continuous oxygen. Patients who received nocturnal oxygen therapy alone had a higher mortality at both 12 and 24 months (40.8% at 24 months) compared with those who received continuous oxygen therapy (22.4% at 24 months).
The second trial, published in 1981, is the British Medical Research Council Domiciliary (BMRCD) trial.26 In this trial, 87 patients with hypoxemia and COPD were randomized to receive either no oxygen therapy or at least 15 h/d of supplemental oxygen. Similar to the NOTT trial, mortality was significantly higher in the no-oxygen group.
From these two trials, in patients with COPD and hypoxemia, we can infer that supplemental oxygen for >15 h/d is better than no oxygen at all, and that continuous supplemental oxygen is better than nocturnal-only therapy. The entry criteria for these trials form the basis of the current Medicare criteria and ATS guidelines for LTOT. Based on the BMRCD trial, most prescriptions for oxygen are written for at least 15 h/d. The data from the two trials have been extrapolated to other conditions complicated by chronic hypoxemia for which data from large trials are lacking, including cystic fibrosis, restrictive lung diseases, and chronic cardiac diseases.
In patients whose hypoxemia does not meet resting criteria for administration of LTOT, evaluation for exercise-related desaturation may be indicated. Some patients may require supplemental oxygen only with exertion; improved exercise performance may be noted,27 unless cardiac and peripheral circulatory factors are performance-limiting.
Finally, patients with significant desaturation while sleeping may warrant nocturnal supplemental oxygen. Important to note, however, is that in the absence of underlying lung disease, these patients often have obstructive sleep apnea or obesity hypoventilation syndrome, for which noninvasive positive pressure ventilation constitutes primary therapy.28 Table 144-5 lists the physiologic indications for oxygen therapy during sleep.
TABLE 144-5 Physiologic Indications for Nocturnal Oxygen Therapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


