Other Signal Transduction Agents
Alex A. Adjei
Cancer represents a multitude of diseases characterized by uncontrolled cellular proliferation and is the second most common cause of death in Western society.1 Despite advances in diagnosis and treatment, overall survival (OS) remains poor.2 Radiotherapy and chemotherapy are relatively nonspecific, affecting normal cells as well as rapidly proliferating tumor cells,3 and cause toxicities that limit their long-term utility. Tumor responses from cytotoxic chemotherapy are usually transient and unpredictable.4 Furthermore, resistance to chemotherapy is common in all malignancies.
Novel targeted agents that represent improvements over traditional treatments are therefore needed. These agents would ideally have high specificity toward tumor cells, resulting in minimal side effects.5 Increased understanding of molecular mechanisms underlying tumor growth, progression, and metastasis over the past decade have identified promising anticancer targets; in particular, protein kinases—enzymes that modify cellular proteins by catalyzing the transfer of phosphate from adenosine triphosphate (ATP) to either serine/threonine or tyrosine amino acid residues.6 Approximately 90 tyrosine kinases, of the transmembrane receptor type or the cytoplasmic nonreceptor type,7 which regulate pivotal signaling pathways that control normal cellular function and development,6,8 have been identified. Their activity is normally tightly controlled and highly regulated.7 Dysregulated kinase activity, resulting from oncogenic gene mutation or overexpression, has a major role in carcinogenesis.6 For example, overactivation of these enzymes increases tumor cell proliferation and growth, induces antiapoptotic effects, and promotes angiogenesis and metastasis.4
In this chapter, we discuss the discovery, biology, signaling pathways, and pharmacology of protein kinase targets (Fig. 50.1) as a well as a few nonkinase targets involved in cell proliferation, metastasis, and apoptosis. These targets include receptor tyrosine kinases (RTKs) with roles in proliferation—insulin-like growth factor receptor 1 (IGFR-1), mesenchymal-epithelial transition factor (c-Met), c-Kit, Flt-3, and RET (rearranged during transfection); and cytoplasmic nonreceptor kinases involved in proliferation and/or prevention of apoptosis—the serine/threonine kinases Raf, MEK, mammalian target of rapamycin (mTOR) and Aurora kinases, and tyrosine kinases Bcr-Abl and Src.
The kinases involved in angiogenic signaling as well as the epidermal growth factor receptor (EGFR) signaling are discussed elsewhere in this volume.
PROLIFERATIVE RECEPTOR TYROSINE KINASES
IGFR/PI3-Kinase Pathway The phosphatidyl-inositol 3 kinase (PI3K) pathway is one of the most important oncogenic pathways, with estimates suggesting that activating mutations in the p110α catalytic subunit occurs in up to 30% of all human cancer, although this is uncommon in non-small cell lung cancer (NSCLC). The most common mechanism of its aberrant activation though is the loss of the regulatory phosphatase and tensin homologue (PTEN) lipid phosphatase, which occurs frequently in NSCLC. PI3K is activated by Ras and RTKs such as EGFR, c-met, and IGF receptor pathway. Autocrine production of IGF as well as overexpression of its cognate receptor, IGF-1R, are well documented in NSCLC, with downstream signaling chiefly mediated by the PI3K-Akt pathway.
Regardless of the mechanism of its dysregulation, Akt is the chief mediator of downstream signaling through various targets. One key target is the mTOR, a serine/threonine kinase that serves as a key component regulating transcriptional and translational proteins that control cell growth, angiogenesis, apoptosis, as well as amino acid and glucose metabolism. This pathway has been implicated in mediating resistance to cytotoxic chemotherapy as well as resistance to novel agents such as the EGFR tyrosine kinase inhibitors (EGFR TKIs).
IGF Receptor The IGFs help regulate normal cell metabolism, growth, proliferation, differentiation, cell-cell and cell-matrix adhesion, and survival.9 The IGF-1R is expressed in most cells, has ligand-activated tyrosine kinase activity,10 and
activates several downstream signaling pathways, including Raf/MEK/extracellular signal-regulated kinase (ERK).9 IGF bioactivity is influenced by a family of six IGF-binding proteins (IGFBPs) that sequester IGFs, thereby preventing excessive cell growth and/or promoting apoptosis.11 Increased circulating levels of IGF, or an increased ratio of IGF to IGFBP, are implicated in the development of several types of tumors, including breast,12 prostate,13 lung,14 and colon carcinomas.15 Several monoclonal antibodies (MoAbs) and small molecule inhibitors of IGF-1R are in clinical testing.
activates several downstream signaling pathways, including Raf/MEK/extracellular signal-regulated kinase (ERK).9 IGF bioactivity is influenced by a family of six IGF-binding proteins (IGFBPs) that sequester IGFs, thereby preventing excessive cell growth and/or promoting apoptosis.11 Increased circulating levels of IGF, or an increased ratio of IGF to IGFBP, are implicated in the development of several types of tumors, including breast,12 prostate,13 lung,14 and colon carcinomas.15 Several monoclonal antibodies (MoAbs) and small molecule inhibitors of IGF-1R are in clinical testing.
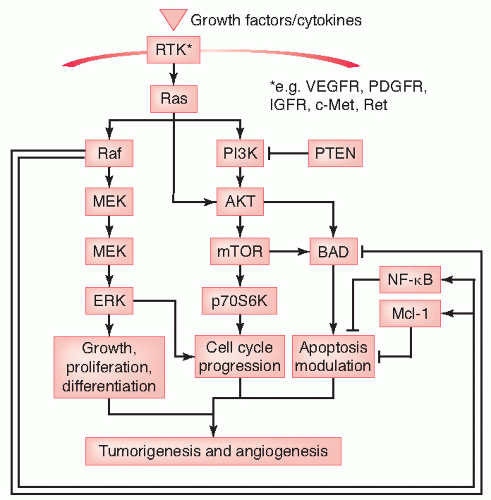 FIGURE 50.1 Overview of the key protein kinase signaling pathways involved in tumorigenesis. AKT, AKT8 virus oncogene homologue; BAD, Bcl2-antagonist of cell death; c-Met, growth factor receptor c-met; ERK, extracellular signal regulated kinase; IGFR, insulin-like growth factor 1 receptor; Mcl-1, myeloid cell leukemia sequence 1; MEK, MAPK/Erk kinase; mTOR, mechanistic target of rapamycin; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; PDGFR, platelet-derived growth factor receptor; PI3K, phosphatidylinositol 3-kinase; PTEN, phosphatase and tensin homologue; Raf, root abundant factor; Ras, ras protein; Ret, ret oncogene; RTK, receptor tyrosine kinase; VEGFR, vascular endothelial growth factor receptor. |
CP-751,871 is a fully humanized IgG2 MoAb antagonist of IGF-1R with preclinical anticancer activity.16 The MoAb interrupts the binding of IGF-I to IGF-1R, IGF-1R autophosphorylation and induces downregulation of IGF-1R in vitro and in tumor xenograft models. In a phase I study, CP-751,871 was administered intravenously every 21 days in advanced solid tumor patients.17 CP-751,871 was escalated to the maximally feasible dose (based on duration of infusion) of 20 mg/kg without any dose-limiting toxicities (DLTs). The correlative studies revealed increased expression of serum insulin and human growth factor hormone, presumably, through a negative feedback loop. The most common adverse events were hyperglycemia, anorexia, nausea, elevated liver transaminases, hyperuricemia, and fatigue. In an exploratory assay, IGF-1R-expressing circulating cancer cells (CTCs) were analyzed. Three patients with detectable IGF-1R expressing CTCs at baseline were reported to have decreased level of CTCs following CP-751,871 administration that rebounded at the end of the 21-day period.17,18 Preliminary phase II results of a randomized first-line advanced NSCLC phase II study of paclitaxel and carboplatin plus/minus CP751,871 were presented at American Society of Clinical Oncology (ASCO) 2007. A total of 46% of patients in the experimental arm achieved objective responses (22/48 patients) versus 32% (8/25 patients) in the control arm.19 An unplanned subgroup analysis by histology has suggested a greater benefit in patients with squamous histology within this trial, but the mature results with additional patient numbers remain to be published. Based on these promising phase studies of CP-751,871 in combination with paclitaxel and carboplatin, phase III studies comparing this combination with chemotherapy alone as frontline therapy in NSCLC has commenced. In addition, a phase III trial of erlotinib with or without CP-751,871 is also ongoing, as are studies in multiple myeloma and other tumor types.20,21,22,23
AMG-479 is a fully humanized anti-IGF-1R MoAb with broad preclinical antitumor activity. The agent showed potent inhibition of the PI3k/Akt axis with increased antitumor effect when combined with anti-EGFR therapies in pancreatic cancer xenograft models.24 In phase I testing of AMG-479, 16 patients with advanced solid tumors received escalating dose of the agent intravenously.25 At 20 mg/kg every 2 weeks, one patient experienced grade 3 dose-limiting thrombocytopenia. No greater than grade 2 hyperglycemia was observed. The agent is currently being tested in non-Hodgkin lymphoma, Ewing sarcoma, and desmoplastic small round cell tumors.26,27 Phase I studies in combination with gemcitabine or panitumumab, as well as phase I/II studies in combination with irinotecan and panitumumab in colorectal cancer are being planned at the time of writing.28
Other anti-IGF-1R agents under phase I/II evaluation include the MoAbs IMC-A12, R-1507, and BIIB022. As well as the oral small molecule inhibitors, XL-288, OSI-906, and nordihydrohuareacetic acid are also being tested.
mTOR mTOR is a serine/threonine kinase, acting downstream of PI3K/AKT.29 The PI3K pathway is activated by Ras, which is often overstimulated in tumors where it contributes to cell cycle progression, inhibits apoptosis, and increases metastatic potential.30 mTOR promotes RTK-induced cell growth, proliferation, and prolongs cell survival via its target, P70S6 kinase, which binds to mitochondrial membranes and inactivates the pro-apoptotic molecule Bcl2-antagonist of cell death (BAD).31
Overactivation of mTOR can also arise through inactivating mutations of the tumor suppressor PTEN gene, resulting in overstimulation of the PI3K/AKT signaling pathway. Loss of the PTEN gene is associated with poor prognosis, resistance to chemotherapy, and various solid tumors, including
glioblastoma multiforme,32 melanoma,33 thyroid,33 breast,34 ovarian, and prostate carcinoma.29 Dysregulation of mTOR signaling is also important in hematologic malignancies, including mantle cell lymphoma.35
glioblastoma multiforme,32 melanoma,33 thyroid,33 breast,34 ovarian, and prostate carcinoma.29 Dysregulation of mTOR signaling is also important in hematologic malignancies, including mantle cell lymphoma.35
The mTOR protein was discovered in the 1990s when the mechanism of action of rapamycin was investigated.36 Rapamycin (sirolimus) is a macrolide isolated from Streptomyces hygroscopicus, a bacterial species native to Easter Island, and has been used widely as an immunosuppressant in organ transplantation.37 Rapamycin has been evaluated orally as an anticancer agent in solid tumors and pancreatic cancer.38,39 mTOR complexes with raptor (regulatory-associated protein of mTOR) and rictor (rapamycin-insensitive companion of mTOR) to form mTOR complex-1 (mTORC1) and mTORC2, respectively. mTORC1 is downstream to Akt and is susceptible to inhibition by rapamycin and its analogs, whereas mTORC2 is an upstream regulator of Akt and the activity is upregulated in certain circumstances as a compensatory response to mTORC1 inhibition.40,41 Interestingly, recent evidence refuted the belief that mTORC2 is rapamycin resistant. It has been demonstrated that mTORC2 can, in fact, be inhibited by rapamycin and its analogs in a time-and cell line-dependent manner.42,43
Temsirolimus (CCI779) is a water-soluble synthetic rapamycin ester available in oral and intravenous formulations.44 The drug is the first of the class to receive Food and Drug Administration (FDA) approval for the treatment of poor-risk untreated advanced renal cell carcinoma (RCC) patients.45,46 In the pivotal randomized trial, temsirolimus was superior to interferon-α alone or in combination with interferon-α in prolonging survival. The most common grade 3 or 4 toxicities were asthenia, anemia, and dyspnea. The recommended dose of temsirolimus for this indication is 25 mg weekly intravenous administration. The drug is currently under testing in various tumor types either alone or in combination therapy.46,47,48,49 A cohort of patients with NSCLC treated with temsirolimus as first-line therapy was assessed in a phase II study conducted by the North Central Cancer Treatment Group (trial 0323).50 In a report of 50 evaluable patients primarily with stage IV disease, partial response (PRs) were observed in four patients (8%) and stable disease for a minimum of 56 days was observed in 15 patients (30%), suggesting an overall clinical benefit rate of 38%. Median progression-free survival (PFS) and OS were 2.3 and 6.6 months, respectively. Temsirolimus was noted to be well tolerated in this population of patients. A phase II study assessed the effect of temsirolimus alone as consolidation treatment in patients with extensive-stage small cell lung cancer (SCLC) in complete remission.51 Temsirolimus was administered until the time of progression and in 85 evaluated patients, the median OS was 8 months. These results have prompted several studies of temsirolimus in combination with chemotherapy in SCLC.
Everolimus (RAD001) is an oral mTOR inhibitor with similar antineoplastic activity as other rapalogs.52,53 In a phase I study in solid tumor patients, the optimal biologic dose for everolimus was determined as 20 mg weekly. This dose achieved pharmacokinetic and pharmacodynamic changes correlated with antineoplastic effects in animal models. Toxicities at this dose were mild and include anorexia, fatigue, rash, mucositis, headache, hyperlipidemia, and gastrointestinal disturbances. 54 When administered on a daily continuous manner, everolimus was well tolerated at a 10-mg dose in patients with refractory or relapsed hematological malignancies. No DLTs were reported and activity was seen in patients with myelodysplastic syndrome.55 The dose of 5 mg/m2 was the maximum tolerated dose (MTD) in pediatric solid tumor patients and DLTs (included diarrhea, mucositis, and elevation of alanine transaminase).56 No objective tumor responses were observed. Everolimus was further tested in a clinical trial exclusively enrolling patients with NSCLC.57 Patients in this phase II trial had an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of ≤2 and had failed ≤2 cycles of platinumbased therapy (arm 1) or ≤2 cycles of platinum-based therapy with an EGFR antagonist (arm 2). Patients were treated with everolimus at a dose of 10 mg daily. In a preliminary report including 85 patients (42 patients on arm 1; 43 patients on arm 2), everolimus was administered as first-or second-line therapy in 64.3% of patients. With response data available for 74 patients in arms 1 and 2, a best response of PR was experienced in 5.3% and 2.8% of patients, respectively. Median PFS was 11.3 and 9.7 weeks on arms 1 and 2, respectively. The most frequent adverse events observed in the study were stomatitis/mucositis, cough, and dyspnea. Although therapy with everolimus was generally well tolerated, the limited antitumor activity led to discontinuation of the study.
A separate phase II study examined the efficacy of everolimus in the setting of refractory SCLC.58 Patients enrolled in this trial had relapsed disease after 1 to 2 previous regimens, no evidence of brain metastases, and ECOG PS of ≤2. Everolimus was administered at a dose of 10 mg daily until disease progression or unacceptable toxicity. At an interim analysis in which 16 evaluable patients had been assessed, three patients had stable disease as a best response, whereas 81% of patients had progressive disease. Similar to the outcome in the aforementioned trial in patients with NSCLC, everolimus was well tolerated but showed limited efficacy in the setting of SCLC. The agent is being tested in non-small cell lung, kidney, prostate, colorectal, and breast cancers as either single-agent or combination therapies.59,60,61,62,63,64
Deforolimus (AP-23573) is the other mTOR inhibitor currently under clinical testing.65,66,67 During phase I testing, the MTD was 18.75 mg/day and mouth sores was the DLT. Antitumor activity was seen in NSCLC, carcinosarcoma, RCC, and Ewing sarcoma. Phase II studies in sarcoma are ongoing.
Combination of Motor Inhibitors with Other Agents for Treatment of Lung Cancer A phase I study of gefitinib and everolimus therapy in 10 enrolled patients resulted in 2 DLT (grade 5 hypotension and grade 3 stomatitis), and 2 PRs were observed in 8 evaluable patients.68 A phase II trial in previous smokers with stage IIIB/IV NSCLC69 enrolled 2 cohorts of patients including previously untreated patients and patients with previous therapy with a platinating agent and
docetaxel. Partial responses were observed in 4 of 23 evaluable patients (17%). This observed response rate in a group of smokers, for whom gefitinib has lessened antitumor activity, was encouraging. Frequently encountered toxicities with the regimen included diarrhea, rash, and mucosal ulcerations.
docetaxel. Partial responses were observed in 4 of 23 evaluable patients (17%). This observed response rate in a group of smokers, for whom gefitinib has lessened antitumor activity, was encouraging. Frequently encountered toxicities with the regimen included diarrhea, rash, and mucosal ulcerations.
A phase I clinical trial combining everolimus with erlotinib70 in patients with advanced NSCLC who had failed ≤2 chemotherapy regimens with an ECOG PS of ≤2 revealed 1 complete response and 3 PRs in 38 evaluable patients.
PKC Inhibitors Overexpression and increased activity of protein kinase C-β (PKC-β) have been implicated in transformation and tumorigenesis in NSCLC,71,72,73 and recent evidence suggests a link between the PKC and protein kinase B/AK transforming (AKT) pathways.74,75
Enzastaurin, an oral serine/threonine kinase inhibitor, targets the PKC and AKT pathways and so inhibits phosphorylation of glycogen synthase kinase-3β and S6 kinase.76 In a phase I dose-escalation study of daily oral enzastaurin (20 to 750 mg), four patients with advanced lung cancer had stable disease for 3 to 12 months.77
A phase II trial of enzastaurin to determine the 6-month PFS rate in 55 patients with advanced, metastatic NSCLC did not achieve its primary end point of a 20% PFS rate; however, 13% of the patients had PFS for ≥6 months.78
Akt Inhibitors Akt, also known as protein kinase B (PK-B), is a serine/threonine kinase upstream to mTORC1 and is implicated in the formation and maintenance of malignancies.79 Akt is an attractive target as mTOR because of its role in several important cellular functions, including cell cycle progression, protein translation and transcription, apoptosis, and cellular metabolism. The side effects from Akt inhibition could theoretically be more severe than mTOR, given its key role in the axis, and the fact that its upstream of mTOR. The development of this class of agents has been challenging and disappointing so far.
Perifosine, a lipid-based derivative of miltefosine, is perhaps the best-characterized Akt inhibitor in clinical development now. The compound inhibits Akt translocation to the cell membrane and exhibits in vitro antiproliferative effects in several cancer cell lines.80 It should be noted, however, that perifosine is a relatively nonspecific AKT inhibitor, and that by interrupting with cell membrane biology, pleiotropic effects on several signaling pathways and molecules are seen. Perifosine was tested as a daily oral dose on a 3-week cycle in patients with advanced solid tumors.81 The patients reported dose-dependent gastrointestinal adverse events, such as nausea, diarrhea, and vomiting, which led to early therapy discontinuation in increasing number of patients at higher dose levels. The MTD was determined at 200 mg/day. An alternative loading/maintenance dosing schedule was tested in patients with advanced solid tumors.82 The MTD was a loading dose of 150 mg every 6 hours for 4 doses followed by 100 mg once daily for maintenance. The DLTs during the loading period were nausea, diarrhea, dehydration, and fatigue and were manageable with prophylactic antiemetics. However, the side effects were more difficult to manage during the maintenance period. Despite encouraging evidence in preclinical studies, perifosine failed to demonstrate significant single-agent anticancer activity in sarcoma, melanoma, pancreatic, and head and neck cancers during phase II testings.83,84,85,86,87,88,89,90,91,92,93 Perifosine continues to be evaluated as single-agent or combination therapies. GSK690693 is another Akt inhibitor planned for phase I testing.94 Several lipid-and peptide-based Akt inhibitors are being evaluated preclinically.95
Currently, there is limited clinical experience with inhibitors of PI3k and 3-phosphoinositide-dependent protein kinase-1 (PDK-1). The PI3k inhibitors undergoing phase I evaluation include PI-103, BGT-226, BEZ-235, XL-765, and XL-147.96,97,98,99,100 Current PDK-1 inhibitors are derivatives of staurosporin and celecoxib.95 UCN-01 is a staurosporin derivative that inhibits multiple kinases including PDK-1 and has in vitro pro-apoptotic activity.101,102 The drug is synergistic with cytotoxic agents in preclinical studies but the pro-apoptotic activity seemed to be from the inhibition of checkpoint homologue (Chk1), a cell cycle checkpoint kinase.103 UCN-01 can be administered intravenously as an initial 72-hour continuous infusion on a monthly schedule or short infusion over 3 hours every 28 days with the second and subsequent doses at 50% of the first.104,105 However, the clinical activity of UCN-01 was not associated with PI3k/Akt/mTOR pathway inhibition and its role as a PDK-1 inhibitor remains ambiguous. OSU-03012 is a celecoxib derivative that inhibits PDK-1 and induced apoptosis in rhabdomyosarcoma cell lines.106 This drug is currently still under preclinical evaluation.
c-Met The protein product of c-Met proto-oncogene is a transmembrane RTK that is activated by the multifunctional cytokine hepatocyte growth factor (HGF)/scatter factor (SF),107 which increases signaling through the Raf/MEK/ERK pathway.108 HGF/SF mediates epithelial cell morphogenesis, migration, invasion, and differentiation.107 HGF/SF is produced predominantly by mesenchymal cells, whereas c-Met is expressed primarily in epithelial cells.
Overexpression of c-Met is implicated in several tumor types, including glioblastomas, RCC,109 hepatocellular carcinoma (HCC),110 pancreatic adenocarcinomas,111 thyroid carcinomas,112 melanoma,113 breast,107 gastric,114 pancreatic,115 prostate, and lung cancers.116,117
There are currently several HGF/c-MET inhibitors under clinical evaluation. AMG-102 is a fully humanized immunoglobulin G2 (IgG2) MoAb against HGF with antitumor activity in preclinical models.118 The interim result of the phase I study was reported at American Society of Clinical Oncology (ASCO) annual meeting in 2007.119 The agent was administered intravenously at 0.5-, 1-, 3-, 5-, 10-, or 20-mg/kg dose levels. Patients with advanced solid tumors received a single dose followed by a 4-week treatment free period to assess safety and pharmacokinetic profile. The treatment was subsequently resumed at a 2 weekly schedule. Thirty-one patients were treated at doses up to 20 mg/kg. DLTs include dyspnea/hypoxia (at 0.5-mg/kg dose) and gastrointestinal bleed (at 1-mg/kg dose). Common treatment-related side effects include fatigue, constipation, anorexia, nausea, and vomiting. Pharmacokinetic
analysis showed a linear relationship in the dose range of 0.5- to 20-mg/kg and no anti-AMG-102 antibodies were detected following administration. The 20-mg/kg dose was deemed tolerable and safe and the agent is being tested in RCC and malignant glioma.120,121
analysis showed a linear relationship in the dose range of 0.5- to 20-mg/kg and no anti-AMG-102 antibodies were detected following administration. The 20-mg/kg dose was deemed tolerable and safe and the agent is being tested in RCC and malignant glioma.120,121
XL-880 is an oral small molecule inhibitor of c-MET and has activity against vascular endothelial growth factor receptor 2 (VEGFR2), PDGFR-β, kit, FLT3, Tie-2, and Ron. The interim result of the phase I study was presented at ASCO 2007 and two schedules were tested: a “5-day on/ 9-day off” schedule and a daily fixed-dose schedule.122 Fifty-one solid tumor patients were enrolled and hypertension was universally observed. The DLTs with the first schedule were proteinuria and elevated lipase and liver enzymes. The MTD was 3.6 mg/kg and the MTD for the second schedule was not reached at the time of analysis, and common side effects include hypertension and fatigue. Correlative studies showed inhibition of c-MET, RON, Erk, Akt, and increased apoptosis at dose levels less than MTD. The agent is being evaluated in papillary RCC gastric and head and neck cancers.123,124,125
ARQ-197 and PF-02341066 are similar oral small-molecule c-Met inhibitors that are in early phase trials.126 The recommended phase II dose for ARQ-197 was determined to be 120 mg twice daily. Common side effects include fatigue, diarrhea and constipation, and grade 3 elevated liver enzymes were the more severe toxicity.127 Compounds with activity against the HGF/c-MET axis in the preclinical pipeline include MGCD-265, SU-11274, and MGCD-265.128,129,130,131
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


