Normal Physiology of the Cardiovascular System: Introduction
The principal function of the cardiovascular (CV) system is to deliver oxygen and nutrients to and remove carbon dioxide and wastes from metabolizing tissues. This is accomplished by means of two specialized circulations in series: a low-resistance pulmonary and a high-resistance systemic circulation driven by specialized muscle pumps, the right and left heart (each in turn composed of a thin-walled atrium and thicker-walled ventricle), respectively. Although CV physiology can be understood at a number of hierarchical levels, the complex interplay among the intrinsic properties of the cardiomyocytes and isolated muscle, chamber mechanics, and their modulation by variable cardiac-loading conditions and neurohormonal and renal compensatory mechanisms determines the integrated performance of the CV system. Accordingly, CV physiology will be examined at cellular, isolated muscle, and organ (isolated heart and integrated systems) levels.
Cellular Basis of Contraction
The rhythmic beating of the heart distinguishes it from all other organs. The normal heartbeat is initiated by a complex flow of electrical signals called action potentials. The action potential results from highly coordinated, sequential changes in ion conductances through gated sarcolemmal membrane channels (Fig. 5–1).
FIGURE 5–1.
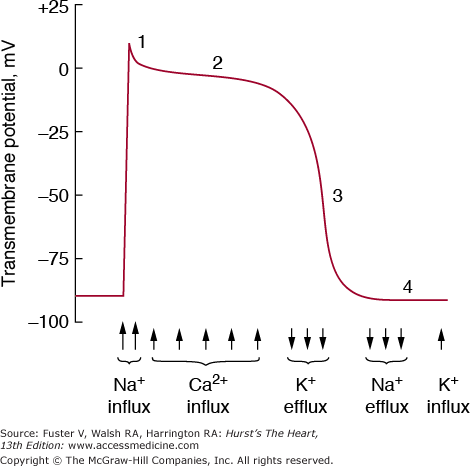
Phases of the action potential and major associated currents in ventricular myocytes. The initial phase 0 spike is not labeled. See discussion of excitation in text. In specialized conduction system tissue, there is spontaneous depolarization during phase IV. Ca2+, calcium; K+, potassium; Na+, sodium. Reproduced with permission from LeWinter MM, Osol G. Normal physiology of the cardiovascular system. In: Fuster V, Alexander RW, O’Rourke RA, Roberts R, et al, eds. Hurst’s The Heart. 11th ed. New York: McGraw-Hill, 2004:87-112.
Increases in transmembrane potential from a resting value of −80 to −90 mV to approximately +30 mV (depolarization) represents phase 0 (the rapid upstroke) of the action potential and results primarily from a sudden increase in sodium (Na+) permeability; this permits a large inward current of Na+ ions to flow down an electrochemical gradient by means of voltage-and time-dependent fast Na+ channels. The upstroke is caused by a regenerative process, that is, depolarization leads to Na+ influx, which leads to further depolarization. The rapid opening of the activation gates for the fast Na+ channel is immediately followed by a slower closing of inactivation gates, which interrupts the influx of Na+ into the cell. The membrane must be fully repolarized for inactivation gates to reopen and conduct another action potential, a process called recovery.
Phase I (the notch) is the initial rapid repolarization phase of the action potential, which is carried by potassium (K+) and to a lesser extent, chloride (Cl−) ion conductance. Phase II of the action potential is unique to cardiac muscle; this plateau phase results from a balance of inward calcium (Ca2+) and outward K+ currents. The slow inward (L-type) Ca2+ channel is activated at threshold potentials above −50 mV, is maximal at approximately 0 to 10 mV, peaks rapidly, and inactivates slowly. Some Na+ channels remain active and carry a late Na+ current throughout the action potential plateau; increased activity of this channel contributes to the abnormal repolarization and increased intracellular calcium in heart failure and ischemic heart disease. Phase III is the final rapid repolarization that restores resting potential and is caused by inactivation of the Ca2+ current and an increase in the outward K+ current. Several ionic K+ pumps contribute to the plateau and repolarization: (1) the inwardly rectifying K+ current (IK1), a K+ conductance that generates the resting potential, turns off during phase 0 and is inactive until repolarization begins—it also generates a small outward current late in repolarization; (2) the transient outward K+ current (ITO), responsible for the initial phase I repolarization; and (3) the delayed outward K+ current (IK), the primary current responsible for initiating final repolarization turns on slowly at the final phase of the action potential. After repolarization, the Na+K+ adenosine triphosphatase (ATPase) pump extrudes accumulated intracellular Na+ and pumps extracellular K+ into the cell. Ionic balance across the sarcolemmal membrane is also maintained by the action of a sodium–calcium exchanger.1
All myocardial cells are excitable, that is, when adequately stimulated, they can generate an action potential. However, only specialized cells are capable of reaching threshold potential and firing without such an outside stimulus (automaticity). Phase IV of the action potential represents the slow, spontaneous diastolic depolarization responsible for the property of automaticity. Normally, action potentials reach threshold potential and depolarize spontaneously and rhythmically only in the primary pacemaker of the heart, the sinoatrial (SA) node. However, cells in other areas (atrial cells near the ostium of the coronary sinus, the distal atrioventricular [AV] node, and the His-Purkinje fibers) are capable of automaticity when not suppressed by the faster firing of the SA node. The slope and maximal diastolic potential of the pacemaker potential and the threshold potential determine the rate of impulse formation; the former is modulated by the autonomic nervous system (sympathetic stimulation increasing the slope of the pacemaker potential and accelerating the rate of firing, and parasympathetic stimulation producing the opposite effects). Several ionic currents, specific for the site of impulse genesis, can be involved in the pacemaker current. In the SA node, an inward Ca2+ current and an outward delayed K+ current that is activated during the plateau and deactivated during phase IV contribute to depolarization. The “funny” current (pacemaker current; IF), which slowly activates on hyperpolarization, is a critical determinant of the slope of diastolic depolarization and is therefore a key regulator of pacemaker activity.2 The T-type Ca2+ channel is present in the developing heart and adult atrium (and ventricular myocytes from hypertrophied and failing hearts), plays a role in the cardiac pacemaker current, and is involved in release of Ca2+ from internal stores.
Effective cell-to-cell communication is essential for rapid, uniform conduction of action potentials and a resultant effective, synchronized myocardial contraction. The organized distribution of local currents that comprise the depolarization wave flow from cell to cell by means of gap junctions. These clusters of transmembrane channels connect the plasma membranes of adjacent myocytes and form low-resistance pathways.3 These channels are composed of two connexons; each connexon is a hexamer of connexins, members of a multigene family of conserved proteins.
The cascade of biological processes that begins with the cardiac action potential and ends with myocyte contraction and relaxation defines cardiac excitation–contraction (E–C) coupling (Fig. 5–2). The E–C coupling is intimately related to calcium homeostasis, myofilament calcium sensitivity, and functions of cytoskeletal and sarcomeric proteins and forms the biophysical underpinnings of the inotropic state of the heart.4 Because E–C coupling is a direct manifestation of myocyte calcium handling, an understanding of the calcium transient and calcium homeostasis is essential.
FIGURE 5–2
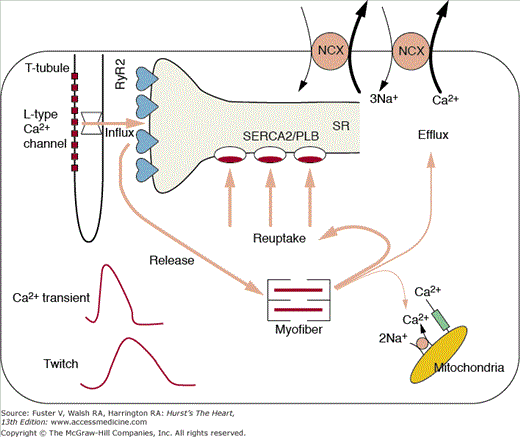
Major components of excitation–contraction coupling (E–C coupling). Influx of calcium is predominantly through the L-type calcium channel. The arrow through the channel denotes the amount of activator calcium and is an index of the E–C coupling gain. The relative magnitudes of calcium release, reuptake, and efflux are denoted by the arrow widths. The resultant calcium transient and muscle twitch are shown in the lower left of the cell. Ca+, calcium; Na+, sodium; RyR2, ryanodine receptor; SERCA2, sarcoplasmic-endoplasmic reticulum calcium ATPase; SR, sarcoplasmic reticulum. Adapted from Scoote M, Poole-Wilson PA, Williams, AJ, et al. The therapeutic potential of new insights into myocardial excitation-contraction coupling. Heart. Apr 89(4):371-376, 2003.
The calcium transient is initiated in response to sarcolemmal depolarization by extracellular calcium (Ca2+) influx through voltage-dependent L-type Ca2+ channels, which instigate the release of stored Ca2+ from the cardiomyocyte endoplasmic reticulum, sarcoplasmic reticulum (SR), via spatially proximate Ca2+ release channels (ryanodine receptor [RyR2]). This latter step, fittingly termed calcium-induced calcium release (CICR), amplifies the amount of calcium available for myofilament binding and force generating actin–myosin crossbridges. Relaxation results from closure of the release channels, resequestration of Ca2+ by the SR Ca2+-ATPase (SERCA2), and crossbridge dissolution. To maintain steady-state calcium homeostasis, the amount of Ca2+ entering the cell with each contraction must be removed before the subsequent contraction. To this end, the Na+-Ca2+ exchanger (NCX) acting in the forward mode competes with SERCA2 for Ca2+ and pumps [Ca2+]i into the extracellular space.
The magnitude of the [Ca2+]i transient modulates the force developed by myofilaments, and factors that modify calcium cycling and/or Ca2+ sensitivity of myofilaments can alter significantly the force and extent of myocyte contraction. The determinants of the cardiac myocyte [Ca2+]i transient are summarized in Table 5–1. Factors responsible for the [Ca2+]i transient amplitude include (1) the calcium current (ICa), primarily caused by Ca2+ influx through the L-type Ca2+ channel, but in small part caused by reverse mode NCX; (2) SR [Ca2+]i content, which determines the amount of releasable calcium; (3) the efficiency of E–C coupling, or the gain (ie, the amount of calcium released by the SR for the calcium current, Δ[Ca2+]i/ICa); and (4) intracellular Ca2+ buffers. The decline of the [Ca2+]i transient is caused by (1) Ca2+ reuptake into SR by SERCA2 (a process modulated by a phosphorylatable regulatory protein termed phospholamban); (2) Ca2+ extrusion from the cell by the NCX; (3) Ca2+ extrusion from the cell by the sarcolemmal Ca2+-ATPase; (4) Ca2+ accumulation by mitochondria; and (5) Ca2+ binding to intracellular buffers (including fluorescent indicators that are used in experimental systems to measure the transient).5
| [Ca2+]i transient magnitude |
| Calcium current (ICa) |
| L-type channels |
| NCX |
| Others (T-type channel, tetrodotoxin sensitive) |
| SR [Ca2+]i content |
| Efficiency of E–C coupling |
| Intracellular Ca2+ buffers |
| [Ca2+]i transient decline |
| SERCA2/phospholamban |
| NCX |
| Sarcolemmal Ca2+-ATPase |
| Mitochondria |
| Intracellular buffers |
Calcium sparks (localized [Ca2+]i transients) are the elementary SR Ca2+ release events that trigger E–C coupling in heart muscle.7 The basis for the generally accepted local control theory of E–C coupling is that Ca2+ sparks are triggered by a local [Ca2+]i established in the region of the RyR2s by the opening of a single L-type Ca2+ channel. The amplitude of Ca2+ sparks is determined by SR Ca2+ load and gating properties of the RyR2. Although the exact nature and origin of Ca2+ sparks are not completely understood, the prevailing view is that the global [Ca2+]i transient is produced by the temporal and spatial summation of a large number of Ca2+ sparks.8 The mechanisms responsible for terminating sparks are not clear, but proteins accessory to the RyR2 (eg, sorcin [FKB12]) have been suggested as playing a key role.9 This calcium signaling system is conceptualized as an integration of subsystems—a “digital” (brief, high concentrations over short distances) and an “analog” (or global intracellular Ca2+) subsystem.6
The sarcolemma is the site where calcium enters and leaves the cell through a distribution of ion channels, transporters, and pumps. The T-tubules are invaginations of the sarcolemma and glycocalyx and are both longitudinal and oblique in their orientation; this system forms a permeability barrier between the cytosol and the extracellular space.7 The membranous surface areas are tissue (atrial cells have poorly developed T-tubules) and species specific. T-tubules play a complex regulatory role in the calcium transient; for example, the ICa is more sensitive to βAR stimulation and intracellular Ca2+ at the T-tubules than at the surface membrane.8 The structural specialization of the sarcolemma include (1) SR coupling in the form of dyads by means of the T-tubule; (2) caveolae, which are invaginations of the sarcolemma that increase surface area and form a scaffold for signaling molecules such as nitric oxide (NO) synthase and protein kinase C (PKC); and (3) the intercalated disc, which takes the form of a gap junction, intermediate junction, or desmosome. Ankyrins are sarcolemmal adaptor proteins that are implicated in the proper expression and membrane localization of ion channels, transporters, dystrophin, and other proteins.
The SR is an intracellular membrane-bounded compartment comprised of terminal, longitudinal, and corbular components (Fig. 5–3). The free walls of the terminal cisternae are apposed to the walls of the T-tubules and form the dyadic cleft; the RyR2 receptors are located in the walls of the terminal cisternae (feet) and face the dyadic cleft. Longitudinal SR is fairly homogenous and contains primarily the SR Ca2+-ATPase proteins, SERCA2, and the associated phosphoprotein phospholamban. In its dephosphorylated state, phospholamban is an endogenous inhibitor of SERCA2. Phosphorylation by PKA (at amino acid serine 16) and calcium-calmodulin kinase II (CaMKII) (at threonine 17) lowers the Michaelis constant (Km) of sarcoplasmic endoplasmic reticulum calcium ATPase (SERCA) and results in enhanced calcium uptake. SR calcium is transported from the tubular lumen of the SR to the terminal cisternae, where it is stored mostly bound to calsequestrin, a low-affinity, high-capacity, calcium-binding protein. Calsequestrin forms a complex with the proteins junctin, triadin, and RyR2. Junctional SR does not come into contact with the sarcolemma; corbular SR is a form of junctional SR that contains calsequestrin and RyR2s but is not coupled to the well-recognized calcium cycling events.9
FIGURE 5–3

Organizational differences between skeletal and cardiac T-tubule junction. The upper panels are side views of the junction (trapezoids are ryanodine receptors [RyRs], and filled ovals are the dihydropyridine receptors [DHPRs]). The lower panels are views from inside the T-tubule at the junction. Note that DHPRs are sparse and less aligned in the heart. RyR, ryanodine receptor; SR, sarcoplasmic reticulum; TT, T-tubules. Reproduced with permission from Bers.9
Myofilaments comprise the contractile machinery of the cell and occupy 45% to 60% of the ventricular myocyte volume (Fig. 5–4). The fundamental unit of the myofilament is the sarcomere, bounded by Z lines on each end, from which the thin actin filaments extend toward the center. At the center of the thick myosin filament is the M line, where the thick filaments are interconnected by M protein and myosin. Titin runs from the M line to the Z line in association with myosin and myosin-binding protein C (MyBP-C); this large structural sarcomeric protein acts as a scaffold for myosin deposition, stabilizes the thick filament, functions as a molecular spring, and plays a critical role in determining the passive stiffness of the heart. Changes in titin stiffness occur during cardiac development and disease states through shifts in the relative expression of the compliant N2BA and stiff N2B titin isoforms. Acute changes in titin stiffness (and as a result, diastolic ventricular function) is produced by PKC and PKG-mediated phosphorylation of an I band–specific domain of titin.10 The functions of MyBP-C are not entirely clear; it may play a role in regulation of contraction by limiting the reach of myosin heads toward the thin filament. It may also play a prominent role in myofilament calcium sensitivity, crossbridge cycling rate, and length-dependent activation. Mutations in the MyBP-C gene are responsible for a significant proportion of familial hypertrophic cardiomyopathy.11 The Z lines are the sites of anchor for cytoskeletal intermediate filaments and actin filaments at the intercalated disks and at focal adhesions. The two major structural complexes involved in the connections between sarcomeric proteins and the extracellular matrix (ECM) are the membrane-spanning integrin complex and the dystrophin complex, which links actin to laminin and collagen.
FIGURE 5–4


Electron micrograph (A) and schematic diagram (B) of a sarcomere. The darkly staining regions that flank the sarcomere are the Z lines. Myosin-containing thick filaments are in the center of the sarcomere and interact with actin-containing thin filaments by way of myosin heads that protrude from the thick filaments. Thin-filament regulatory proteins, the troponin and tropomyosin, provide calcium regulation of the actin–myosin interface. Thin filaments are anchored to the Z line, which is enriched in proteins such as α-actinin and Cap Z. (Right) Membrane complexes that concentrate over Z lines. The dystrophin–glycoprotein complex. Reproduced with permission from McNally E. The cytoskeleton. In: Walsh RA, ed. Molecular Mechanisms of Cardiac Hypertrophy and Failure. London: Taylor and Francis; 2005:309-321.
The myosin molecule consists of two heavy chains with a globular head, a long α-helical tail, and four myosin light chains (Fig. 5–5). The myosin head forms crossbridges with the thin actin filament through an actin-binding domain. Transduction of chemical to mechanical energy and work is the function of myosin ATPase, located in the myosin heads. Myosin heavy chain (MHC) exists as two isoforms, α (fast ATPase and crossbridge formation) and β (slow ATPase and crossbridge formation). In higher mammals, including humans, the β-myosin isoform predominates, but in small mammals, such as mice and rats, the α form is dominant. The most accepted model of energy transduction is the sliding filament theory based on the formation and dissociation of crossbridges between the myosin head and the thin filament that transition through different energetic states. Two myosin light chains (the alkali or essential light chain [MLC1] and the phosphorylatable or regulatory light chain [MLC2]), are associated with each myosin head and confer stability to the thick filament. Phosphorylation of the myofilament regulatory protein troponin modulates the activity of myosin ATPase. Although phosphorylation of MLC2 by myosin light-chain kinase (MLCK) is critical for smooth muscle cell contraction (see the following section), its physiological significance in cardiac muscle (increased calcium sensitivity and rate of force development) is controversial.
FIGURE 5–5
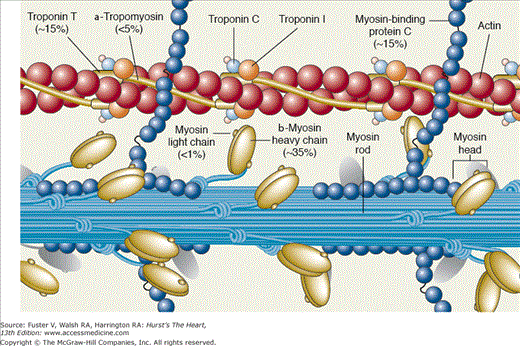
The sarcomeric proteins. Titin, which runs from the M line to the Z line in association with myosin and myosin-binding protein C, is not shown. Reproduced with permission from LeWinter MM, Osol G. Normal physiology of the cardiovascular system. In: Fuster V, Alexander RW, O’Rourke RA, et al, eds. Hurst’s The Heart. 11th ed. New York: McGraw-Hill; 2004:87-112.
The backbone of thin filament is helical double-stranded actin. Tropomyosin is a long, flexible, double-stranded (largely α-helix) protein that lies in the groove between the actin strands and inhibits the interaction between actin and myosin (see Fig. 5–5). The troponin complex is composed of a calcium-binding subunit, troponin C (TnC); an inhibitory subunit that binds to actin, troponin I (TnI); and a tropomyosin-binding subunit, troponin T (TnT), which is attached to tropomyosin. In the resting state, when [Ca2+]i is low, calcium-binding sites of TnC are unoccupied, and TnI preferentially binds to actin; this favors a configuration in which the troponin–tropomyosin complex sterically hinders myosin–actin interaction. In this configuration, crossbridges are in both detached and weakly attached non–force-producing states. When [Ca2+]i rises, calcium binds to the calcium-specific sites on TnC and strengthens the interaction of TnC and TnI; TnI then dissociates from actin, and a conformational change removes the steric hindrance to myosin–actin interaction. Strong binding of actin to myosin begins when the actin–myosin inhibition is relieved. Binding Ca2+ to troponin causes the process of crossbridge formation to spread down the actin filament, and by means of adenosine triphosphate (ATP) hydrolysis, transitions are made from detached or weakly bound states to force-producing states. Release of conformational energy leads to rotation of the myosin head that propels the thin filament along the thick filament. Usually the systolic [Ca2+]i only submaximally activates muscle; the steep relation between [Ca2+] and tension is thought to result from both nearest neighbor interaction and strong actin–myosin binding, which allows for contractile reserve with modest changes in [Ca2+]i. Although this is the most accepted model, other potential explanations exist; all models incorporate the concept that myofilaments are dynamically involved in their state of activation and not simply subject to passive changes in [Ca2+]i. A simplified mechanical model of crossbridge formation is presented in Fig. 5–6.
FIGURE 5–6
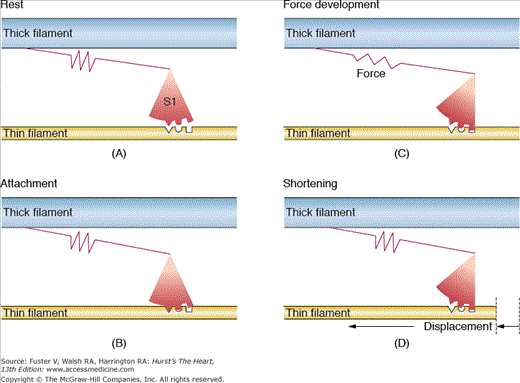
A mechanical model of the crossbridge cycle. A. Detached crossbridge. B. Crossbridge before developing force. C. Attached crossbridge developing force stored in the elastic component. D. Crossbridge rotated and translated so the filaments slide relative to one another. Each step in the cycle can be related to energetically different chemical states. Reproduced with permission from Bers.9
Mitochondria comprise approximately 35% of ventricular myocyte volume and according to their cellular location are designated as either subsarcolemmal or interfibrillar. Mitochondria are the sites of oxidative phosphorylation and ATP generation. Although they have the capacity to buffer large amounts of Ca2+ and are a potential source of activator calcium, classical teaching is that their contribution to E–C coupling is minimal in view of the short time constants involved; variation in mitochondrial Ca2+ during a twitch is imperceptible and thus plays a very minor role in beat-to-beat changes in calcium homeostasis. However, the kinetics of mitochondrial Ca2+ uptake during E–C coupling have recently become controversial.12 Nevertheless, slower increases in mitochondrial Ca2+ content are important with respect to mitochondrial function and energetics; for example, the matrix enzymes pyruvate dehydrogenase, nicotinamide adenine dinucleotide (NAD)–dependent isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase are activated by low [Ca2+]. In addition, the ability to accumulate large amounts of Ca2+ under pathological conditions (eg, ischemia) can help protect against myocyte Ca2+ overload; however, Ca2+ accumulation by mitochondria ultimately slows ATP production.
NO is produced by the myocardium and regulates cardiac function through both vascular-dependent and -independent effects.13 NO has a modest positive inotropic effect on basal contractility in isolated myocytes and the isolated perfused heart but a negative inotropic effect in vivo,14 possibly because of nitrosylation of ion channels responsible for E–C coupling (eg, L-type channel, RyR2). The negative inotropic effects on β-adrenergic stimulated contractility are greater and less controversial and can comprise a critical component of negative feedback over contractile reserve. NO’s positive effects on relaxation or lusitropy (and in part, for negative inotropic effects) are likely to be caused by cyclic 3′, 5′-guanosine monophosphate (cGMP)-mediated reduction in myofilament Ca2+ sensitivity.15 Finally, mitochondrial NO reduces maximal venous oxygen (MVO2) consumption and increases mechanical efficiency (stroke work/MVO2), suggesting that NO regulates energy production as well influencing consumption.
The effects of NO on E–C coupling are confusing and controversial because of the presence of three nitrous oxide synthase (NOS) isoforms that are spatially localized to highly controlled microdomains and linked to disparate signaling pathways and effectors. For example, NOS type III (NOS3) is compartmentalized to the sarcolemmal and T-tubule caveolae, associated with the L-type channel, inactivated by the scaffolding protein caveolin-3, and activated by Ca2+/calmodulin and Akt phosphorylation. NOS3 produces its negative inotropic and positive lusitropic effects by means of cGMP activation.
In contrast, NOS type I (NOS1), which is also activated by Ca2+/calmodulin and can be inactivated by caveolin-3, is localized to cardiac SR and is involved with calcium homeostasis. NOS1 increases the open probability of the cardiac RyR2 and modulates β-adrenergic mechanics, calcium transients, and the force–frequency relationship, although the mechanisms remain controversial. Nevertheless, accumulating data suggest that NO plays an important role in E–C coupling vis-á-vis modulation of Ca2+ channel activity, myofilament Ca2+ sensitivity, and mitochondrial respiration.14
Heart rate dependence of cardiac contractility reflects basic cycling kinetics of calcium and is critically dependent on SR function. Processes related to force–interval behavior (eg, mechanical restitution, force–frequency, postextrasystolic potentiation [PESP]) are important insofar as they represent fundamental physiological control mechanisms, they are used as indices of myocardial function, and they play a role in the response to exercise and the development and maintenance of heart failure. Non–steady-state aspects of E–C coupling provide the basis for these phenomena.
Mechanical restitution is the relative refractory period that immediately follows a contraction and is usually explained by the recovery of the RyR2 receptors (because ICa and SR Ca2+ content recover rapidly).16 Mechanical restitution is the basis for PESP, the strong contraction following a weaker extrasystole, because lower [Ca2+] on the extrasystole results in increased ICa (less Ca2+-induced inactivation of the L-channel), less Ca2+ efflux from NCX, and increased SR Ca2+ loading on the postextrasystolic beat. The result is a greater amount of released Ca2+ and therefore a stronger contraction. In the intact heart, the effect of changing preload (and the impact on Frank-Starling and calcium sensitivity) is an important additional mechanism. PESP contributes to the beat-to-beat variability of the pulse in atrial fibrillation. Mechanical alternans, the alternating contraction amplitude at a constant heart rate that is seen in heart failure, is explained by a similar interplay of RyR2 refractoriness (which is increased in heart failure), ICa in-activation, NCX competition, and SR Ca2+ load.9
The relationship between pacing rate and force (force–frequency relationship) can be understood similarly by these non–steady-state phenomena. Increased pacing rate overcomes the encroachment on mechanical restitution and produces an increase in force because of rate-dependent increases in ICa, INa (which results in less Ca2+ efflux by the NCX), diastolic [Ca2+]i (less time for efflux and greater influx/second), releasable SR Ca2+ content, and fractional SR Ca2+ release.17 A phenomenon similar to the force–frequency relationship is observed when the effects of heart rate on the time constant of isovolumic relaxation are examined. Thus, similar to the effect on contraction, relaxation is augmented at higher rates of stimulation.
An emerging concept is that the molecular machinery of E–C coupling is involved in the long-term regulation of gene expression by a process known as excitation–transcription (E–T) coupling. Despite periodic oscillations of [Ca2+] from 100 nM to 1 μM during E–C coupling, transcription regulatory proteins (eg, NFκB, janus N-terminal kinase [JNK], nuclear factor of activated T cells [NFAT]) are calcium activated. The amplitude and duration of the calcium signal; the presence of microdomains and anchoring proteins; and linkages through calmodulin, kinases, and phosphatases are important mechanisms for discriminating important regulatory cues and resolving this apparent paradox.18 For example, in adult ventricular myocytes, inositol 1,4,5 triphosphate (InsP3) receptors localized to the nuclear envelope are involved in the local control of Ca2+ (so-called reactive signaling) for a CaMKII-mediated activation and regulation of a histone deacetylase.19
CaMKII regulates proteins involved in calcium transport; ion channels; and cell contraction, metabolism, and proliferation by phosphorylation. Phosphorylation substrates for CaMKII that are involved in modulating contraction–relaxation include phospholamban (PLB), SERCA2a, L-type Ca2+ channels, and the RyR2.20 CaMKII phosphorylates the transcription factor cyclic adenosine monophosphate (cAMP) response element binding (CREB), which promotes transcription of cfos.21 In addition, CaMKII has autoregulatory properties that are dependent on the frequency of Ca2+ spikes, a process thought to have a role in neuronal memory. Little is known about in vivo CaMKII activation, but biochemical data suggest that that CaMK might be primed to respond to Ca2+ spikes. Thus, calcium-dependent regulation by calmodulin and CaMKII has both acute responses affecting E–C coupling and chronic responses that influence the expression levels of proteins involved in E–C coupling.4 In vascular smooth muscle cells, the L-type channel (via the RhoA/ROK pathway) and the calcineurin/NFAT pathway are involved in the regulation of cell differentiation.22
Arterial smooth muscle cells exist in the partially constricted state. The principal determinant of vascular tone is membrane potential, which is achieved through activation of voltage-gated calcium channels. There is a steep relation between [Ca2+] and vascular tone, and therefore membrane potential must be highly regulated to maintain appropriate vascular resistance. The resting potential of smooth muscle cells ranges from −40 to −70 mV, lower than cardiac muscle because of greater Na+ permeability. Thus, the rising phase of the action potential is produced by inward calcium current through the slow L-type Ca2+ channels. Contraction results directly from depolarization-induced Ca2+ influx and indirectly by means of CICR-activation of the contractile apparatus. Relaxation results from lowering cellular Ca2+ via Ca2+ ATPase pumps and hyperpolarization of the cell by activation of K+ channels.
A distinctive feature in smooth muscle is that Ca2+ acts as a second messenger to activate MLCK, which phosphorylates the myosin light chains and produces force. Ca2+ binds to calmodulin, and this complex activates MLCK. Phosphorylation of the 20-kD light chain stimulates actin-activated myosin ATP hydrolysis and contraction. Relaxation occurs when there is dissociation of Ca2+ from calmodulin, inactivation of MLCK, and dephosphorylation of myosin by myosin light-chain phosphatase (Fig. 5–7).
FIGURE 5–7
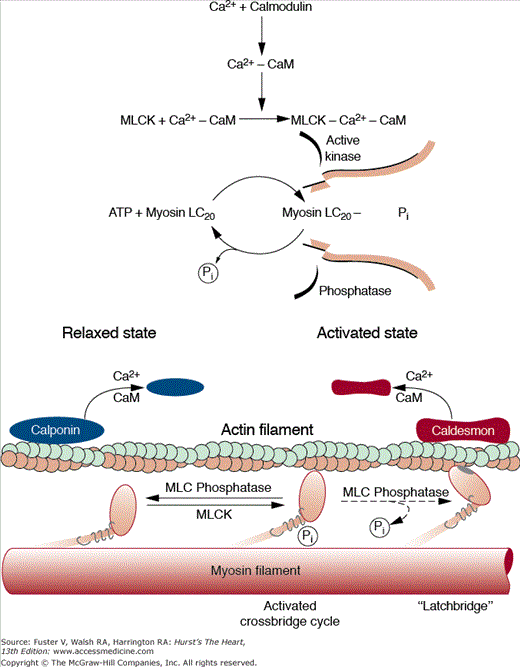
The molecular basis of regulation of smooth muscle contraction. Stimulation of muscarinic receptors increases the [Ca]i because of entry of external calcium (Ca2+) and release of Ca2+ from internal stores. Ca2+ binds to calmodulin (CaM) and the Ca2+–CaM complex subsequently binds to and activates myosin light-chain kinase (MLCK). Phosphorylation of myosin by MLCK stimulates actin-activated myosin-ATP hydrolysis, which produces contraction. Relaxation begins with the cessation of agonist stimulation, resulting in decreased [Ca]i, dissociation of Ca2+ from CaM, inactivation of MLCK because of dissociation of CaM, dephosphorylation of myosin by phosphoprotein phosphatases, and relaxation. ATP, adenosine triphosphate. Reproduced with permission from Paul R, Heiny JA, Ferguson DG, Solaro RJ. Diversity of muscle. In: Sperelakis N, Banks RO, eds. Essentials of Basic Science: Physiology. 2nd ed. Boston, MA: Little, Brown and Company; 1996:217-225.
Unlike myocardial cells, both cAMP and cGMP inhibit the activity of the slow Ca2+ channels. Thus, both NO (which increases cGMP) and β-adrenergic agonists (which increase cAMP) are vasodilators. Stimulation of delayed rectifier channels and sarcolemmal Ca2+ pumps produce vasodilation. Angiotensin II and α-agonists cause vasoconstriction by phospholipase C (PLC)–mediated production of inositol trisphosphate (IP3; which releases Ca2+) and diacylglycerol (DAG), which stimulates PKC phosphorylation of the Ca2+ channel and inhibition of the delayed rectifier channel.
Properties of Myocardial Contraction
Fundamental to cardiac muscle function are the relationships between force and muscle length, velocity of shortening, calcium, and heart rate. The maximal force developed at any sarcomere length is determined by the degree of overlap of thick and thin filaments and therefore the number of available crossbridges. Force increases linearly until a sarcomere length with maximal overlap (~2.2 μm) is achieved (Fig. 5–8), beyond which force and overlap gradually declines to zero (ie, the descending limb). The descending limb of the length–tension relationship is prevented by the strong parallel elastic component in cardiac muscle. The ascending limb of the length–tension relationship (equivalent to the Frank-Starling relationship that relates preload to cardiac performance) is also caused by a length-dependent increase in myofilament calcium sensitivity (Fig. 5–9A). This has been explained by enhanced calcium binding to TnC, narrower interfilament gaps at long sarcomere length, and increased SR calcium release and uptake at longer sarcomere lengths.
FIGURE 5–8
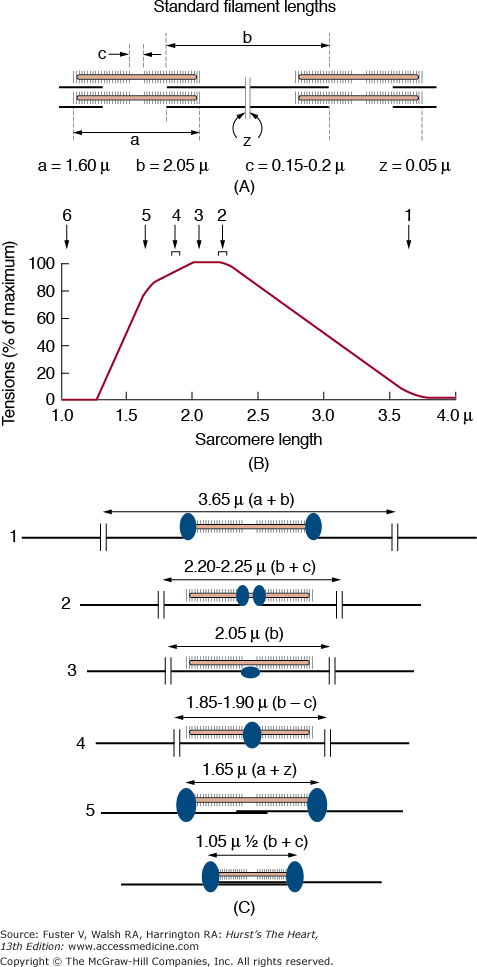
Structural basis for the active isometric force–length relationship. A. Sarcomere Z line to Z line minimal sarcomere length. B. Optimal sarcomere length with maximal cross bridge overlap. C. H line with no possible cross bridge attachment. From Gordon AM, Huxley AF, Julian FJ. The variation in isometric tension with sarcomere length in vertebrate muscle fibres. J Physiol. 1966;184(1):170-192.
FIGURE 5–9

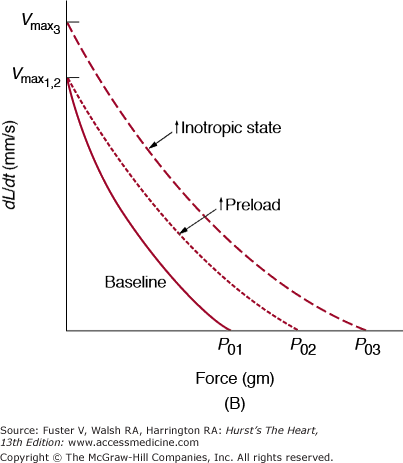
A. Length–shortening and length–velocity relationships from isotonic contractions at a constant afterload. As muscle length (preload) increases, shortening (ΔL) and velocity of shortening (dL/dt) increase. An increased inotropic state shifts the curve upward and to the left; conversely, a decreased inotropic state shifts the curve downward and to the right. B. Force–velocity relationship from variably afterloaded contractions. Increased preload causes an increase in the maximum isometric tension (P01 to P02) without a change in the extrapolated velocity of an unloaded contraction (Vmax). An increase in the inotropic state increases P0 and Vmax (dL/dt = velocity of contraction). Reproduced with permission from Hoit BD, Walsh RA. Determinants of left ventricular performance and cardiac output. In: Sperelakis N, Banks RO, eds. Essentials of Physiology. 2nd ed. Boston, MA: Little, Brown and Company; 1996:269-278.
The relationship between force and velocity of contraction is hyperbolic (Fig. 5–9B); at maximum force (isometric force), shortening cannot occur, and at zero force (ie, unloaded muscle), velocity is at a maximum (Vmax), reflecting the maximum turnover rate of myosin ATPase. Therefore, alterations in the myosin isoform (ie, α, fast; β, slow) such as those seen in response to pressure overload, have an effect on Vmax.
Another fundamental property of cardiac muscle is the force pCa2+ relation. Shorter sarcomere lengths decrease Ca2+ sensitivity, and caffeine and various inotropic drugs (eg, levosimendan) are potent calcium sensitizers. β-Adrenergic stimulation results in a cAMP-dependent phosphorylation of cardiac TnI and a resultant decrease in myofilament calcium sensitivity; thus, for a positive β-adrenergic receptor (βAR)–inotropic effect, the amplitude of the calcium transient must more than compensate for reduced βAR-mediated myofilament sensitivity.9
The final property relates heart rate to contraction and relaxation. Increasing the heart rate increases contractility; this is related to the Ca2+ capacity and load of the SR. A related phenomenon, frequency-dependent acceleration of relaxation (FDAR), results from CaMKII phosphorylation of phospholamban (or by some other mechanism that increases SR Ca2+ transport).23 CaMKII might be activated by the increased [Ca2+]i that occurs with increased stimulation rates; however, the precise mechanisms are unresolved. The physiologic implications for faster relaxation at increased heart rates, when the diastolic filling periods are shortened, are discussed in the following section.
When a strip of heart muscle is attached at both ends so that the length is fixed and then electrically stimulated, the muscle develops force without shortening (Fig. 5–10A). A fundamental property of striated muscle is that the strength of this isometric twitch is dependent on the initial resting muscle length, or preload (Fig. 5–10B). As cardiac muscle is stretched passively, the resting tension rapidly rises and prevents overstretching of the sarcomeres. If additional load is applied before contraction (ie, the preload), stimulation causes contraction with an increased peak tension and rate of tension development (dT/dt). Thus, total tension includes both active and passive tension. The length–tension relationship, which forms the basis for the Frank-Starling relationship, is depicted in Fig. 5–10C. The inotropic state is defined operationally as a change in the rate or extent of force development that occurs independently of the loading conditions. The biophysical basis of the inotropic state includes the subcellular processes that regulate myocyte cytosolic calcium and actin–myosin crossbridge cycling. In isolated cardiac muscle, changes in the inotropic state are measured by changes in the peak isometric tension and dT/dt at a fixed preload.
FIGURE 5–10
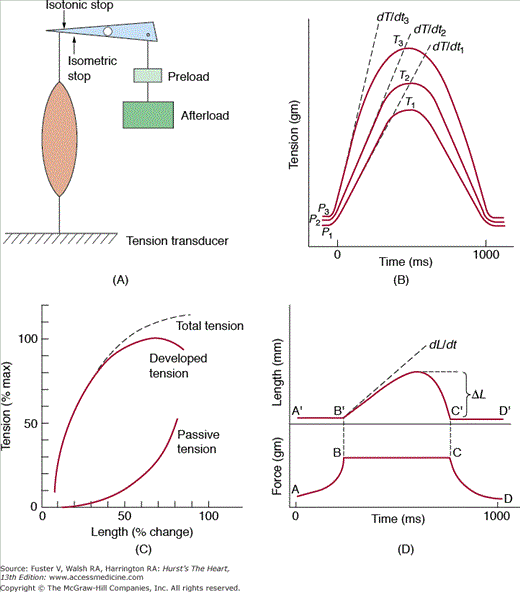
Contractions in isolated muscle. A. Isolated muscle preparation. Muscle is attached to a level arm at one end and fixed to a tension transducer at the other. The muscle is stretched by applying a weight (preload) at one end of the lever arm. A stop prevents muscle shortening. B. Tension–time curves of isometric twitches at three levels of preload. With increased preload, peak tension (T) and the maximum rate of tension development (dT/dt) are increased. The time to peak tension is unchanged. C. Length–total tension relationship and its components, passive and active tension. As muscle is stretched, the absolute passive and total tension increase. D. Superimposed tension–time and length–time recordings from afterloaded isotonic contractions. After preload is applied, a stop is placed to prevent further stretching. Afterload is added, and the muscle is stimulated. Muscle shortens when generated tension equals total load (preload and afterload). Measures of shortening in the isotonic contraction include total shortening (DL) and the initial velocity of contraction (dL/dt). Redrawn with permission from Ross J. Best & Taylor’s Physiological Basis of Medical Practice. 12th ed. Baltimore, MD: Williams & Wilkins; 1990.
If isolated cardiac muscle is allowed to shorten, the contraction is termed isotonic (Fig. 5–10D). Initial muscle length is determined by applying a preload; an additional load known as the afterload
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


