Chapter 34
Normal Coagulation
Kenneth G. Mann, Kathleen E. Brummel-Ziedins
Maintenance of blood flow is essential to life. Blood flow may be lost because of pump failure, leakage of blood, or blood vessel obstruction. The coagulation system has evolved to deal with the last two events by providing qualities that maintain blood in a fluid state within the vasculature. This challenge is met by a three-component system that promotes vascular blood fluidity while anticipating the requirement for blockade of extravascular blood flow. The system involves vascular endothelial cells, blood, and extravascular tissue. These three compartments maintain fluidity or produce an integrated response to attenuate blood leakage by localized clotting at the site of vascular injury, with the dimensions of the response in proportion to the injury.
The processes of blood coagulation and fibrinolysis are the primary defense systems of the vasculature. The opposing forces of fibrin clot formation and dissolution maintain hemostasis and preserve vascular function and integrity. Procoagulant events that culminate in generation of α-thrombin and formation of a fibrin clot protect the vasculature from perforating injury and excessive blood loss. Fibrinolysis removes the clot and initiates mechanisms involved in tissue repair and regeneration. Hemostasis therefore refers to multiple discrete processes that center on α-thrombin generation, fibrin clot formation, and fibrin clot dissolution.
Circulating and adherent cells and circulating and cell membrane–associated proteins carry out key roles in the coagulation and fibrinolytic pathways. Hemostasis is not a passive state but instead is actively maintained by the vascular system. Specific cellular and protein interactions are required to maintain a state of equilibrium. When the system is perturbed, an integrated series of processes are required to initiate procoagulant events and to promote fibrinolysis and tissue repair. Each individual process that contributes to hemostasis must operate properly or the entire system is compromised. A balance among procoagulant, anticoagulant, and fibrinolytic factors is required to prevent uncontrolled bleeding or, conversely, excessive clot formation.1
Many of the processes involved in hemostasis are not well understood. Epidemiologic studies have expanded our knowledge about key factors that determine the risk of cardiovascular disease. However, cardiovascular disease is still the primary cause of death in the United States and western Europe. To more effectively treat and ultimately prevent cardiovascular events, we continue to examine the processes that contribute to blood coagulation and fibrinolysis. In the following sections, we summarize the current concepts governing the roles of protein components: their structures, functions, and regulation. This is followed by descriptions of their dynamic relationships and techniques for monitoring of these processes.
Procoagulant, Anticoagulant, and Fibrinolytic Proteins, Inhibitors, and Receptors
History and Nomenclature
Current knowledge of the components involved in the complex process of blood coagulation (Fig. 34-1) is the result of observations that date to the second century, when it was noted that if two brothers died of bleeding after circumcision, the third must not be circumcised.2 During the centuries, many hypotheses were envisioned about the transformation of fluid blood to a gel-like mass as it escaped the body.3 These included speculations that blood clotted because it cooled on exposure to air or that it dried as it left the body.4 The realization that clots stem blood loss did not occur until the beginning of the 18th century,3,4 and it was not until the 19th century that the existence of thrombin, the key enzyme in blood coagulation, was recognized.3,5
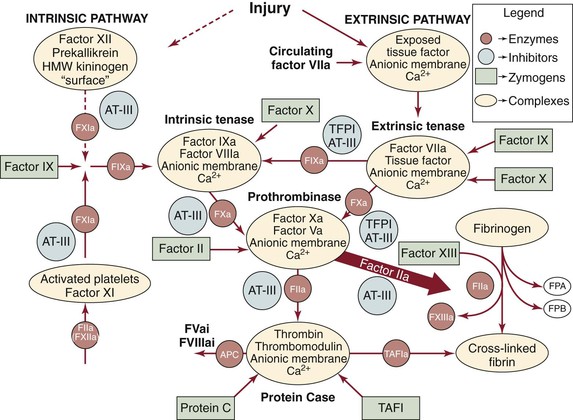
Figure 34-1 Overview of hemostasis. Two pathways can be used to initiate coagulation: the primary extrinsic pathway (shown on the right) and the accessory pathway (historically called the contact or intrinsic pathway, shown on the left). AT, Antithrombin; HWM, high-molecular-weight; TAFI, thrombin-activatable fibrinolysis inhibitor; TFPI, tissue factor pathway inhibitor. (Modified from Brummel-Ziedins K, et al: Chapter 21 In Lee GR, et al, editors: Wintrobe’s clinical hematology, ed 11, Philadelphia, 2003, Lippincott Williams & Wilkins.)
In 1905, Paul Morawitz hypothesized that in the presence of Ca2+ and “thromboplastin,” prothrombin was converted to thrombin, which in turn converted fibrinogen to the fibrin clot.6 Four clotting factors were identified: factor I, fibrinogen; factor II, prothrombin; factor III, thromboplastin; and factor IV, calcium ion. As more coagulation factors were identified, initially by bleeding pathology and subsequently by laboratory clotting tests,7,8 they were assigned consecutive Roman numerals, with activated forms distinguished by an a after the Roman numeral designation.
To describe the multiple simultaneous processes involved in generation of a hemostatic response, we identify an inventory of the procoagulant, anticoagulant, and fibrinolytic participants in blood coagulation. Subsequently we describe the connections between these components and the dynamics of the process.
Vitamin K–Dependent Proteins
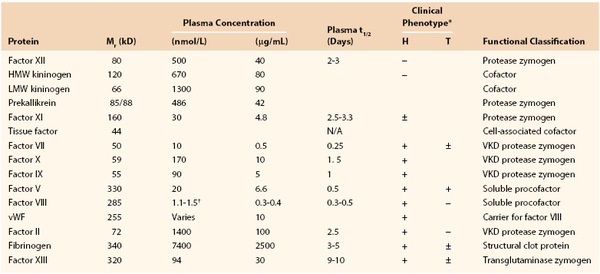
The vitamin K–dependent proteins, synthesized in the liver, play central roles through either procoagulant or anticoagulant functions. The family includes the procoagulant factors VII, IX, X and prothrombin and the anticoagulants protein C, protein S, and protein Z (Fig. 34-2 and Tables 34-1 and 34-2). Except for protein S and protein Z, these proteins in their active forms are serine proteases related to trypsin. Cleavage of specific peptide bonds converts the vitamin K–dependent zymogens to their active serine protease forms. All share noncatalytic domains, each of which is characterized by highly conserved regions that provide specific binding characteristics essential for their function. The domain organizations of the vitamin K–dependent proteins are illustrated in Figure 34-2. (For reviews on vitamin K–dependent proteins, see references 9 to 13).
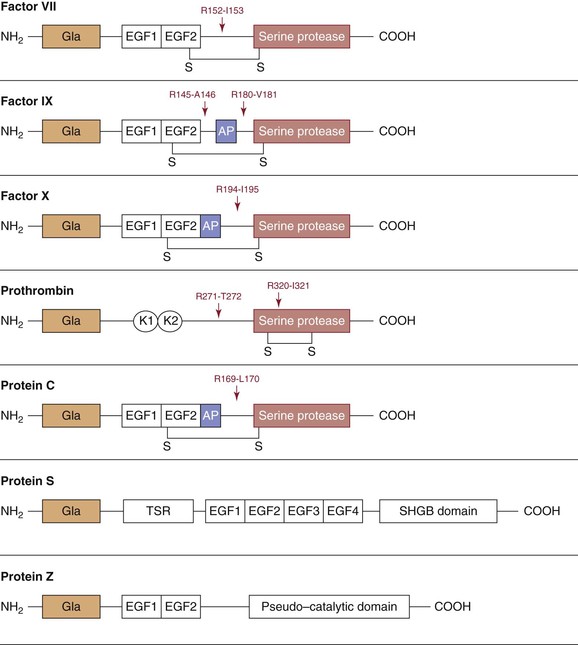
Figure 34-2 Schematic representation of the vitamin K–dependent proteins. The building blocks for these proteins include an NH2-terminal Gla domain with 9 to 13 γ-carboxyglutamate residues followed by either an epidermal growth factor (EGF)–like domain in factor VII, factor IX, factor X, protein C, protein S, and protein Z or kringle (K) domains in prothrombin. Activation cleavage sites, disulfide bonds (-S-S-), and activation peptides (AP) are illustrated. The sex hormone–binding globulin (SHBG) and thrombin-sensitive regions (TSR) are also represented in proteins.
Vitamin K is essential for the biosynthesis of these clotting factors by participating in a cyclic oxidation and reduction process that converts 9 to 13 amino-terminal glutamate residues into γ-carboxyglutamate residues (Gla) (for review, see references 12, 14, and 15). This posttranslational modification enables the vitamin K–dependent proteins to interact with Ca2+ and membranes.16,17 Inhibition of the Gla residue modification is the basis for “blood-thinning” anticoagulant therapy with coumarin derivatives (e.g., warfarin), which are chemically similar in structure to vitamin K and interfere with the redox cycle. The level of anticoagulation achieved with vitamin K antagonist therapy in individuals taking the same dose regimen is variable.18,19 Altered sensitivity to warfarin has been identified in patients when it is prescribed after surgery.20 Factors affecting the efficacy of treatment include liver function in the synthesis of clotting factors, influence of other medications, and dietary intake and absorption of vitamin K.21 Therefore, monitoring of oral anticoagulant therapy with the prothrombin time is essential.18,19 New specific, active site–directed synthetic oral anticoagulants that do not require monitoring have recently been approved (i.e., rivaroxaban, dabigatran).22
NH2-terminal Gla domains are followed by either a Kringle domain in factor II or an epidermal growth factor like–domain in factor VII, factor IX, factor X, protein C, protein S, and protein Z (see Fig. 34-2). Protein S is not a serine protease precursor but instead contains a thrombin-sensitive region before the epidermal growth factor domain and a sex hormone–binding globulin–like domain in the COOH terminus.23 Protein Z contains a “pseudo–catalytic domain” in the COOH terminus and is not a zymogen.24
Cofactor Proteins
Cofactor proteins (Fig. 34-3) either circulate in plasma (factor V and factor VIII) or are the cell-bound tissue factor (TF) and thrombomodulin (TM).
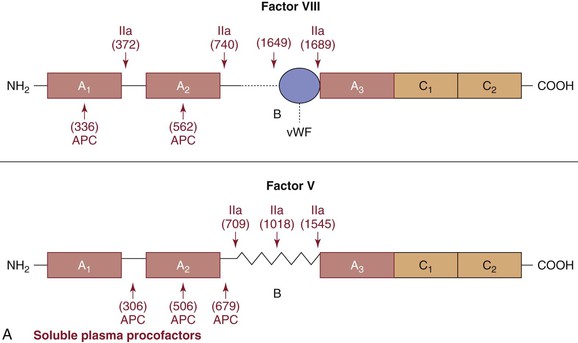
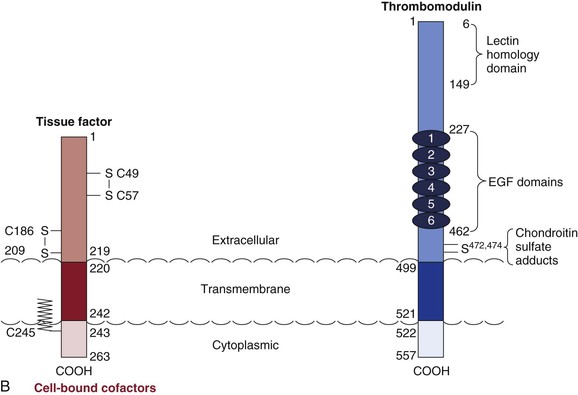
Figure 34-3 Cofactors. A, Plasma procofactors factor VIII and factor V. The linear domain structures (A1-A2-A3-C1-C2) are represented. APC, Activated protein C; vWF, von Willebrand factor. B, Cell-bound cofactors tissue factor and thrombomodulin. Epidermal growth factor (EGF), transmembrane, and cytoplasmic domains are illustrated.
Soluble Plasma Procofactors
Factor V.
Factor V is a large single-chain glycoprotein25–28 that circulates in plasma (see Fig. 34-3A and Table 34-1) and in the alpha granules of platelets; the 18% to 25% of the total factor V present in platelets29 is secreted on platelet activation. Factor V is cleaved at several sites by α-thrombin to the cofactor factor Va.30 The cleavage sites shown in Figure 34-3A excise the B region and provide a two-chain factor Va molecule that functions as a factor Xa receptor and a positive modulator of factor Xa catalytic potential in the prothrombinase complex.31 Factor Va is proteolytically inactivated by activated protein C (APC)32,33 with cleavage sites shown in Figure 34-3A. Cleavage at R506 reduces activity, whereas the slower cleavage at R306 eliminates activity. The importance of this regulatory mechanism is illustrated by the “APC resistance” syndrome associated with factor V Leiden.34 Individuals with factor V Leiden have a G-to-A substitution at nucleotide 1691 in the factor V gene that results in an Arg506→Gln mutation.35 Factor Va Leiden has normal cofactor activity in the prothrombinase complex. However, factor Va Leiden is more slowly inactivated because the Arg506→Gln mutation eliminates the faster cleavage site. A review is available of factor V in hemostasis.31
Factor VIII.
The procofactor factor VIII, or antihemophilic factor, circulates in plasma in complex with the large multimeric protein von Willebrand factor (vWF; see Fig. 34-3A).36 The molecule is synthesized as a single chain but proteolyzed at a variety of sites in the B chain and secreted as a two-chain molecule.37 Factor VIII is activated by α-thrombin cleavage at three sites (Arg372, Arg740, and Arg1689) to generate the heterotrimeric cofactor factor VIIIa.38 The vWF binding site at the NH2 terminus of the light chain is removed from the factor VIII protein through cleavage by α-thrombin at Arg1689. Factor VIIIa forms a complex with the serine protease factor IXa, Ca2+, and a membrane, which results in the “intrinsic” factor tenase. Factor VIIIa function is downregulated by the spontaneous dissociation of the noncovalently associated A2 subunit produced by inactivation and by APC. Factor VIII is homologous (40% identity) with factor V.39
Deficiency of factor VIII, or hemophilia A, is a well-characterized bleeding disorder linked to the X chromosome. Severe hemophilia A therefore occurs almost exclusively in males with a frequency of 1 in 5000 to 10,000.40
Cell-Bound Cofactors
Tissue Factor.
TF is a transmembrane protein that functions as a nonenzymatic cofactor with factor VIIa in the extrinsic tenase complex (Fig. 34-4).41 TF is not expressed in blood in the absence of injury or inflammatory stimulus. Presentation of TF to blood triggers the primary “extrinsic” pathway for hemorrhage control (see Fig. 34-1).42–44 There are no known deficiencies of human TF, and absence of TF in mice is lethal during embryonic development.45 The most commonly accepted sources of functional TF are the subendothelium exposed after vascular damage and monocytes stimulated by cytokines; however, there is also controversy about functional TF circulating in blood.46–50
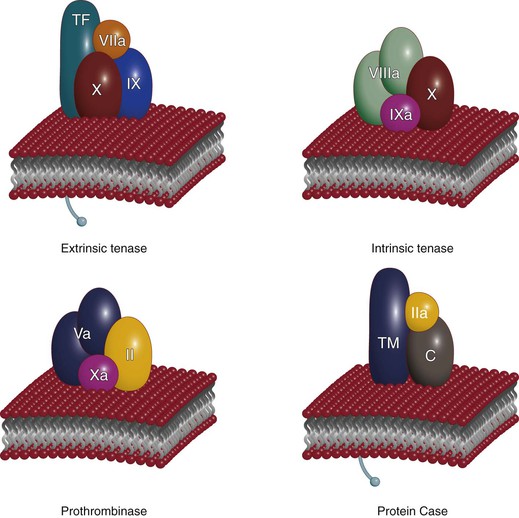
Figure 34-4 Vitamin K–dependent complexes. Three procoagulant complexes (extrinsic tenase, intrinsic tenase, and prothrombinase) and one anticoagulant complex (protein Case) are illustrated. Each serine protease is shown in association with the appropriate cofactor protein and zymogen substrate or substrates on the membrane surface. Calcium ion is not shown. TF, Tissue factor; TM, thrombomodulin. (Redrawn from Mann KG: Coagulation explosion, Vermont Business Graphics, 1997.)
TF is composed of an extracellular domain, a transmembrane domain, and a cytoplasmic domain. Two disulfide bonds (S-S) are identified in the extracellular domain, and the cytoplasmic cysteine (C245) is found in a thiol ester linkage to a fatty acid (see Fig. 34-3B). The extracellular cysteines have been suggested to provide regulatory functions.
Thrombomodulin.
TM is a type 1 transmembrane protein constitutively expressed on the surface of vascular endothelial cells (see Fig. 34-3B). Five distinct domains can be identified in TM: a lectin-like domain, a domain containing six epidermal growth factor–like regions, a small extracellular domain rich in threonine and serine residue (two, S472 and S474, as sites of chondroitin sulfate adducts), a membrane-spanning region, and a cytoplasmic tail.
TM is a high-affinity receptor for α-thrombin and meizothrombin and acts as a cofactor for the activation of protein C.51 The endothelial cell protein C receptor provides cell-specific binding sites for both protein C and APC.52,53 Once α-thrombin is bound to TM, its procoagulant activities (e.g., generate fibrin; activate factor V, factor VIII, factor XI, and platelets) are neutralized and the rate of inactivation of α-thrombin by antithrombin is increased.54–56 Conversely, meizothrombin, an intermediate in α-thrombin formation by cleavage at R320 (see Fig. 34-2), is an efficient protein C activator. APC activation by the thrombin-TM complex leads to inactivation of the cofactors factor Va and factor VIIIa, suppressing thrombin formation (see Fig. 34-2).57 The thrombin-TM (protein Case) complexes also have an antifibrinolytic role through activation of thrombin-activatable fibrinolysis inhibitor (TAFI).58–60
TM activity on the surface of endothelial cells is increased by inflammatory cytokines,61 and this decrease may contribute to hypercoagulation in inflammatory states.
Complexes
Vitamin K–dependent protein complexes are essential for establishing hemostatic balance (see Fig. 34-4). Each complex is composed of a serine protease (factor VIIa, factor IXa, factor Xa, or thrombin [factor IIa]), a cofactor that functions as a receptor/enhancer for the enzyme (factor VIIIa, factor Va, TF, TM), Ca2+, and a negatively charged membrane presented by cells (e.g., endothelial cells, monocytes, platelets). There are four vitamin K–dependent enzyme complexes (see Fig. 34-4): the extrinsic tenase complex (factor VIIa–TF), the intrinsic tenase complex (factor IXa–factor VIIIa), the prothrombinase complex (factor Xa–factor Va), and the anticoagulant protein Case complex (thrombin-TM).
These membrane-bound complexes serve to localize enzymatic activity to the appropriate regional site required for their enzymatic functions. The formation of the protease-cofactor-membrane enzymatic complexes results in the only biologically relevant enzymatic activity for factor VIIa, factor IXa, and factor Xa. The thrombin-TM complex effectively converts thrombin from being a procoagulant to an anticoagulant enzyme.
When the serine protease enzyme is associated with its respective cofactor on an appropriate membrane with Ca2+, the specific reactions occur at enhanced rates, 104– to 109-fold greater than the enzyme-substrate combination in solution.62 The importance of these complexes in the formation of a hemostatic plug is to note that if it takes 4 minutes for blood to clot in a healthy person, in the absence of membrane and cofactor, blood clot formation would take approximately 3.8 years.
Intrinsic (Accessory) Pathway Proteins
The designation intrinsic (accessory) pathway emerged as the relationships between genetic deficiencies and bleeding pathologic processes have been established. Deficiencies of proteins associated with the intrinsic or accessory pathway (factor XII, prekallikrein, and high-molecular-weight kininogen [HMWK]) are not associated with abnormal bleeding, even after surgical challenge.63–65 These proteins were initially found through laboratory clotting tests. In contrast, deficiencies of the protein components of the extrinsic or primary pathway (prothrombin and factors V, VII, VIII, IX, and X) can lead to severe bleeding diatheses.66–71 The physiologic role of the accessory pathway is therefore not understood.72
Factor XI represents an intersection point for the two pathways. Individuals with factor XI deficiency (hemophilia C) express variable bleeding with surgical challenge,73,74 thus establishing a role for factor XI in hemostasis. During the hemostatic process, formation of factor XIa appears to be catalyzed by α-thrombin as part of a positive feedback loop stemming from α-thrombin generation.75 Factor XIa then functions in the propagation phase of α-thrombin generation in association with the primary pathway by activation of factor IX.76
Three proteins, factor XII, prekallikrein, and HMWK, are required for activity of the intrinsic or accessory pathway, which does not require biologic activation. Factor XII and prekallikrein are zymogens that are activated to generate serine proteases, and HMWK is a nonenzymatic procofactor (Fig. 34-5). Activation of this pathway in vitro is accomplished when factor XII autoactivates to factor XIIa with exposure to foreign surfaces, including glass, kaolin, dextran sulfate, and sulfatides.77–79 The substrates for factor XIIa, prekallikrein and factor XI, exist in a noncovalent complex with HMWK and become activated to kallikrein and factor XIa, respectively.80 Positive feedback loops exist in which kallikrein cleaves HMWK, thereby freeing bradykinin and allowing more prekallikrein and factor XI to associate and activate.81 This pathway is also negatively regulated through cleavage of HMWK by factor XIa.82
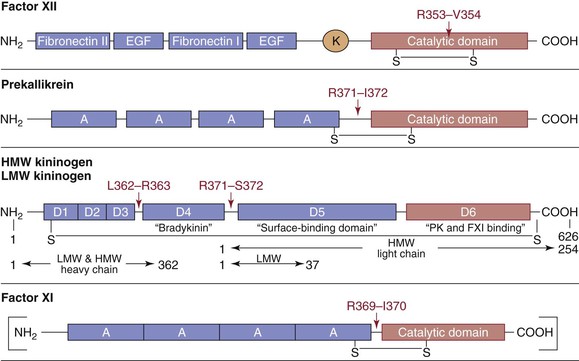
Figure 34-5 Schematic representation of the intrinsic (accessory) pathway proteins. Factor XII, prekallikrein (PK), high-molecular-weight (HMW) kininogen, low-molecular-weight (LMW) kininogen, and factor XI (FXI) are shown with their various domains depicted. A, Apple domain; EGF, epidermal growth factor domain; K, kringle domain. Cleavage sites and key interchain disulfide bonds (S-S) are shown. For the kininogens, horizontal arrows indicate the amino acid residues defining heavy- and light-chain regions of the activated forms of the cofactors. The factor XI molecule is a dimer of the illustrated monomer. Factor XI is a disulfide-linked homodimer with each subunit containing four apple domains, which provide binding sites for high-molecular-weight kininogen, thrombin, factor XIIa, platelets, and heparin. Activation of factor XI (by thrombin or factor XIIa) results in a conformational change that permits factor IX binding to the apple 3 domain.
Although these proteins have no defined role in hemorrhage control, the accessory pathway factors may play roles in disseminated intravascular coagulation associated with systemic inflammatory response syndromes83,84; they may also be involved in the promotion of thrombus stability85,86 and thus may provide targets to control pathologic coagulation.87 Recent evidence suggests that biologic activation of the contact pathway system may be accomplished through assembly of these proteins on endothelial cell membranes and that prekallikrein is activated by an endothelial cell membrane cysteine protease rather than by factor XII.88,89 The accessory pathway is important in cardiopulmonary bypass because of contact between blood components and synthetic surfaces.84
Stoichiometric Inhibitors
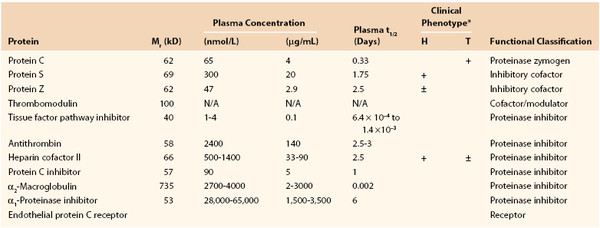
An array of inhibitors is present in blood with both specific and broad-spectrum actions. Inhibitors of clot formation are antithrombin, tissue factor pathway inhibitor (TFPI), heparin cofactor II, and protein C inhibitor (Fig. 34-6). These function to localize, limit, and control coagulation.
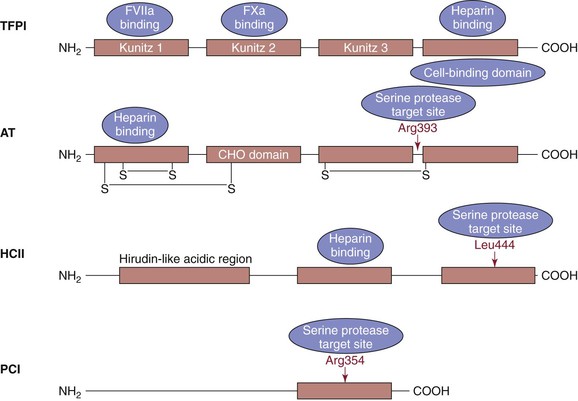
Figure 34-6 Stoichiometric inhibitors. Tissue factor pathway inhibitor (TFPI) has three Kunitz domains (the residues are illustrated below each domain). The Kunitz 1 domain binds factor VIIa, the Kunitz 2 domain binds factor Xa, and the Kunitz 3 domain has been reported to bind protein. The COOH terminus of TFPI contains a basic region, the cell-binding domain that also binds to heparin. Antithrombin (AT) has two intrachain disulfide bonds (-S-S-) at its NH2 terminus and one in its COOH terminus with a carbohydrate-rich domain (CHO) in between. The region of interaction between the active sites of target proteases and AT is called the reactive center loop, R393-S394. Heparin binding occurs in the NH2 terminus. Heparin cofactor II (HCII) contains an NH2-terminal hirudin-like region, a heparin- or dermatan sulfate–binding region, and a reactive center loop. The reactive site is Leu444. Protein C inhibitor (PCI) is a serine protease inhibitor that inhibits several proteases, including activated protein C, thrombin, and factor Xa. It is also a potent inhibitor of the thrombin-thrombomodulin complex. The reactive bond (Arg354) in the reactive center loop is shown.
Antithrombin
Antithrombin is a member of the serpin proteinase family and circulates in blood as a single-chain glycoprotein (see Fig. 34-6).90 Congenital antithrombin deficiency exhibits an autosomal dominant pattern of inheritance, with an incidence of 1 in 2000 to 5000.91 Individuals with this deficiency have partial expression of antithrombin and are prone to thromboembolic disease.92 Complete absence of antithrombin is lethal.
Antithrombin is a broad-spectrum anticoagulant, interacting with most proteases participating in the coagulation cascade (see Fig. 34-1), including α-thrombin, factor Xa, factor IXa, factor VIIa–TF, factor XIa, factor XIIa, plasma kallikrein, and HMWK.93–95 Heparins and heparan sulfates potentiate these reactions and are used in the treatment of thrombosis. When antithrombin is complexed with heparin, its rate of inhibition of several coagulation proteases is accelerated by up to 10,000-fold. The general mechanism of inhibition involves reaction of the active site of the enzyme with a peptide loop structure (the reactive center loop) of antithrombin to form a tight, equimolar (1 : 1) complex. Inactivation proceeds through the formation of a covalent bond between antithrombin and the protease, followed by structural rearrangements of both antithrombin and the protease. Antithrombin inhibits at a slower rate when it is not bound to heparin because the reactive center loop is not completely exposed in the absence of heparin.
Antithrombin also displays antiproliferative and anti-inflammatory properties that primarily derive from its ability to inhibit thrombin. In addition, latent or cleaved forms of antithrombin have antiangiogenic activity.96
Tissue Factor Pathway Inhibitor
TFPI (also called extrinsic pathway inhibitor or lipoprotein-associated coagulation inhibitor) is a multivalent, Kunitz-type inhibitor that circulates in plasma as a heterogeneous collection of partially proteolyzed forms (see Fig. 34-6).97–100 Ninety percent of circulating TFPI is found associated with lipoproteins, primarily low-density lipoprotein.99,101,102 Parenteral TFPI is cleared from the circulation principally by the liver and has an unusually short half-life (minutes) compared with other proteinase inhibitors.
Several reviews of TFPI have been published.103–109 The importance of TFPI in blood coagulation is illustrated with transgenic mice that exhibit complete deficiency (−/−) of TFPI, which is incompatible with birth and survival.110 However, this lethality can be rescued by heterozygous or homozygous factor VII deficiency,111 which implies that diminishing the level of factor VII lessens the need for TFPI-mediated inhibition of the factor VIIa–TF coagulation pathway during embryogenesis.111 Mice bred to have a combined heterozygous TFPI deficiency and homozygous apolipoprotein E deficiency exhibit an increased atherosclerotic burden.112 These observations suggest a role for TFPI in protection from atherosclerosis and as a potential regulator of thrombosis.
TFPI is the principal stoichiometric inhibitor of the extrinsic factor tenase complex (factor VIIa–TF).113 Effective TFPI inhibition of the factor VIIa–TF complex depends on the presence of factor Xa. Thus, inhibition of the extrinsic factor tenase by TFPI occurs only after significant generation of factor Xa. Inhibition by TFPI is achieved by formation of the stable quaternary complex TF–factor VIIa–TFPI–factor Xa and by formation of the factor Xa–TFPI complex.
Heparin Cofactor II
Heparin cofactor II is a member of the serpin family (see Fig. 34-6). Like antithrombin, heparin cofactor II inhibits thrombin in a reaction that is accelerated more than 1000-fold by heparin.114 However, unlike antithrombin, the only coagulation enzyme inhibited by heparin cofactor II appears to be thrombin.115 Heparin cofactor II may play a role in protection from thrombosis during pregnancy. Increased levels of dermatan sulfate in the maternal and fetal circulation116 along with increased levels of heparin cofactor II in pregnant women have been reported.117,118
Protein C Inhibitor
Protein C inhibitor is a serpin also known as plasminogen activator inhibitor-3 (PAI-3). Protein C inhibitor is considered nonspecific in that its targets range from procoagulant (serine proteinases), anticoagulant, and fibrinolytic enzymes to plasma and tissue kallikreins, the sperm protease acrosin, and prostate-specific antigen.119,120 The major target of protein C inhibitor, as its name suggests, is APC.121–123 Because no patients have been documented with a deficiency, the actual function of protein C inhibitor in vivo is yet to be elucidated.
Endothelium and Platelets
Blood cells and the vasculature are crucial to normal hemostasis. Multiple processes involving components of the vessel wall, circulating platelets, and plasma protein moieties interact to maintain blood fluidity. These processes must be precisely choreographed to allow the vasculature to perform myriad complex physiologic activities (Fig. 34-7).
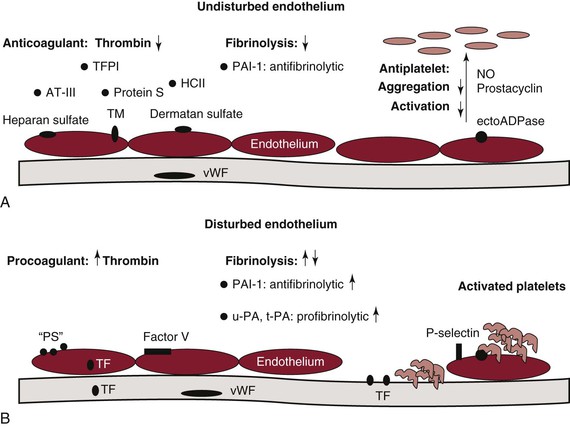
Figure 34-7 The endothelium in hemostasis. A, Under normal conditions and in the absence of injury or chemical stimulus, the undisturbed endothelium actively downregulates thrombin generation through production of tissue factor pathway inhibitor (TFPI), antithrombin (AT), protein S, heparin cofactor II (HCII), heparan sulfate, thrombomodulin (TM), and dermatan sulfate. The undisturbed endothelium is also antifibrinolytic and secretes plasminogen activator inhibitor-1 (PAI-1). In the absence of stimulus, the endothelium is likewise antiplatelet and prevents platelet adhesion activation, secretion, and aggregation through production of nitric oxide (NO), prostacyclin, and the membrane-associated protein ectoADPase. B, When the endothelium is disturbed, the endothelium becomes procoagulant and accelerates thrombin formation by exposing or expressing anionic phospholipid (“PS”), limited tissue factor (TF), and factor V. The fibrinolytic response is modulated by the release of both antifibrinolytic and profibrinolytic molecules. Urokinase (urinary plasminogen activator, u-PA) and tissue plasminogen activator (t-PA) are profibrinolytic and serve to activate plasminogen, whereas PAI-1 inhibits both enzymes and is antifibrinolytic. Platelet activation, secretion, and aggregation are also promoted under conditions in which the endothelium is disrupted. Exposure of von Willebrand factor (vWF) in the subendothelial matrix allows platelets to attach to the surface of the vessel. P-selectin likewise promotes platelet attachment.
The early work of Seegers identified the phospholipid requirements for coagulation.124 The biologic elements contributing to the phospholipid requirements include damaged vascular tissue, activated platelets, and inflammatory cells. These contributions of the membrane to the formation and expression of procoagulant complexes are essential. However, the nature of the membranes that support procoagulant reactions is poorly understood. Mechanically damaged cells can provide the anionic membrane bilayer inner leaflet phospholipids, which can support general procoagulant complex formation, but more subtle cellular activation events also generate selective complex-forming sites on intact cells. Activated platelet membranes express individual binding sites for the factor IXa–factor VIIIa and factor Xa–factor Va complexes. Pathologic hemorrhage is therefore associated with thrombocytopenia and is also seen in a rare disease, Scott’s syndrome, which appears to result from the improper presentation of these platelet binding sites.125
Endothelium
The endothelium plays key roles because of its strategic interface with organs, tissues, and circulating blood. The endothelium varies considerably in morphology and physiologic function in different parts of the vasculature. This complex cellular network not only provides a structural barrier to contain flowing blood but also actively regulates blood pressure, vascular tone, permeability, and processes involving other cells, such as smooth muscle cells, leukocytes, and platelets, and deposits an intricate basement membrane and extracellular matrix.126 In addition, the endothelium is involved in inflammatory and immune responses and angiogenesis.127 Defects in vascular endothelial function therefore have profound physiologic implications. Excessive bleeding can result from structural abnormalities of the endothelial cell layer or supporting matrix. Impaired plasminogen activator inhibitor-1 (PAI-1) expression or secretion (or both) by the endothelium likewise promotes bleeding through increased fibrinolytic activity.128 Conversely, endothelial cells are also involved in mediating processes that lead to the formation of atherosclerotic plaque and thrombotic disease.
It is well established that activated platelets, white blood cells, and microparticles in blood can serve the receptor function for the assembly of the procoagulant complexes.129 Recently, it has been observed that red blood cells, in addition to participating in the blood clotting process through their biophysical effect of causing the margination of platelets at the vessel wall during blood flow, also participate biochemically by expressing phosphatidylserine-like membrane sites that support the assembly of procoagulant complexes.130
Platelets
Platelets are vital to procoagulant events and contribute to the fibrinolytic process as well. Like the endothelium, the undisturbed platelet presents a nonthrombogenic surface. Important components of platelet physiology are surface adhesion protein complexes and platelet secretory granules: alpha granules, lysosomes, and dense granules. The contents of alpha granules include procoagulant and adhesive proteins such as fibrinogen, fibronectin, thrombospondin, vWF, P-selectin, HMWK, platelet factor 4, osteonectin, factor V,29 and factor XI.131 Other alpha granule contents, α1-antitrypsin, protein S, TFPI, and platelet inhibitor of factor XI, are involved in anticoagulant activities.132–134 The alpha granule also contains proteins that meditate both profibrinolytic and antifibrinolytic processes. These proteins include plasminogen, α2-antiplasmin, factor XIII, and PAI-1.135–139 In the unstimulated platelet, the granule contents remain internalized, and anionic phospholipid is sequestered in the inner leaflet of the plasma membrane. The release of prostaglandin I2 and nitric oxide from endothelial cells, the presence of CD39, and the inability of normal plasma vWF to bind spontaneously to the platelet surface are the inhibitory mechanisms that keep platelets unactivated.
When the vascular system is perturbed, platelet plug formation occurs in stages. Initially, platelets adhere and are activated by exposure to collagen, vWF, and other matrix components (Fig. 34-8). The cytoskeleton spreads, platelet-fibrinogen aggregates are formed, and the contents of the granules are secreted.140–142 The activated platelets adhere to each other, endothelial cells, leukocytes, and components of the subendothelial matrix.143 The phosphatidylserine-rich internal face of cell membranes is exposed and presents a highly procoagulant surface to the circulation. In addition, activated platelets express specific receptors or binding sites, or both, for assembly of the procoagulant multiprotein complexes. There are approximately 6000 factor Va binding sites on the activated platelet membrane.144 Factor Va forms part of the receptor for factor Xa. Factor Xa is also reported to bind to effector cell protease receptor-1 molecules expressed on activated platelets.144–147
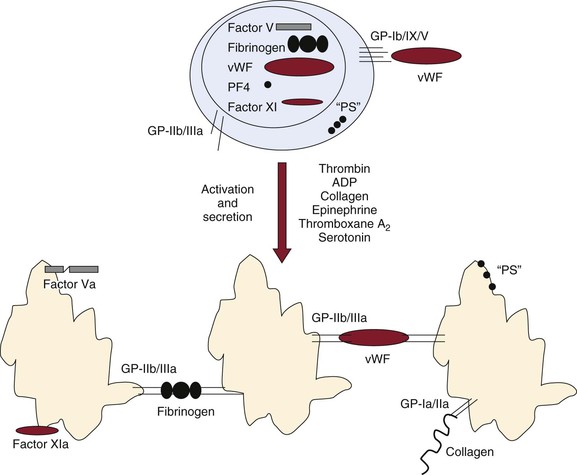
Figure 34-8 Activation, secretion, and aggregation of platelets. Platelets have multiple functions in hemostasis. They serve as reservoirs of factor V, fibrinogen, von Willebrand factor (vWF), platelet factor 4 (PF4), and factor XI. Platelets also contribute a significant portion of the anionic phospholipid (“PS”) necessary for membrane-dependent complex formation and function. In the unstimulated state, proteins and other molecules are sequestered in the platelet granules. Anionic phospholipid is found only in the inner leaflet of the platelet membrane and is not exposed to flowing blood. The glycoprotein Ib/IX/V (GP-Ib/IX/V) complex that recognizes vWF is an active receptor, whereas the GP-IIb/IIIa receptor that recognizes a variety of molecules, including fibrinogen and vWF, is not active. The GP-Ib/IX/V receptor probably allows unstimulated platelets to attach to exposed subendothelial vWF and promotes procoagulant events before platelet activation. On activation by a variety of agonists, platelets secrete granule contents, activate and bind factor V/Va and factor XI/XIa, and expose anionic phospholipid. The GP-IIb/IIIa receptor serves to link platelets to each other and the vessel wall. Collagen receptors such as GP-Ia/IIa promote both platelet activation and aggregation.
α-Thrombin is the most potent activator of platelets, able to overcome most pharmacologic and cytokine inhibitors that depress platelet function. The mechanism by which thrombin activation of platelets occurs is novel and involves two proactivator membrane proteins on the platelet surface, platelet-activated receptors 1 and 4, which when cleaved by thrombin present new NH2 termini that insert themselves into adjacent receptors to trigger platelet activation. Platelet-activated receptor 1 appears to be the more prominent receptor activated by thrombin. Synthetic peptides representing a newly presented membrane-bound NH2-terminus sequence can activate platelets independent of thrombin proteolytic cleavage.
During the extension phase of platelet plug formation, activated platelets accumulate on top of the initial monolayer of platelets bound to collagen. The presence of receptors on the platelet surface allows agonists such as thrombin, adenosine diphosphate, and thromboxane A2 to further recruit circulating platelets into the growing hemostatic plug. Subsequently, during the platelet plug formation perpetuation phase, close contact between platelets promotes growth and stabilization of the hemostatic plug, in part through contact-dependent signaling mechanisms.148
Clot Proteins
The primary proteins of the clot are fibrin and the transglutaminase factor XIIIa. A central event in blood coagulation is the conversion of soluble fibrinogen (factor I) to insoluble fibrin (see Fig. 34-1; for review, see references 149 and 150). Fibrinogen serves as a molecular bridge to support interplatelet aggregation, and it is the precursor of fibrin, which is the main component of the protein scaffolding of the forming hemostatic plug (Fig. 34-9). Platelet aggregation critically depends on fibrinogen binding to activated platelets through the platelet fibrinogen receptor glycoprotein IIb/IIIa (integrin αβ) as well as on fibrin adhesion. Fibrinogen/fibrin also regulates thrombin activity151,152 and thrombin exosite binding to fibrin, which limits the diffusion of thrombin and thereby regulates clot extension.
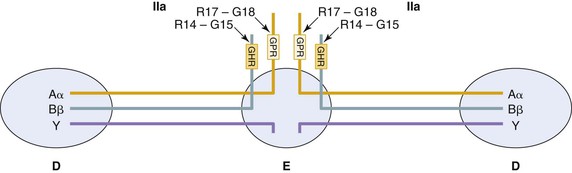
Figure 34-9 A schematic representation of human fibrinogen. Fibrinogen is composed of six polypeptide chains (two Aα, two Bβ, and two γ). These are organized into two identical half-molecules. All six NH2 termini are linked by disulfide bonds in the central or E domain. The three chains extend out from this domain through coiled coils in either direction, terminating in the globular D domains. Thrombin (IIa) cleavage sites at the N termini of the Aα and Bβ (arrows) are indicated, with the new N termini (box) that associate with “holes” in the D domains to form fibrin.
Although it is primarily recognized for its role in hemostasis, fibrinogen is also associated with inflammation. Fibrinogen is an acute-phase reactant, with levels increasing during inflammation. In inflammatory states, fibrinogen functions as a bridging molecule in cell-cell interactions. Fibrin and fibrinogen can modulate a variety of different cell types, including endothelial cells, epithelial cells, leukocytes, platelets, and fibroblasts. Cellular receptors that can bind fibrinogen and fibrin include the integrins αIIbβ3, αVβ3, and α5β1 and the cellular adhesion molecules intercellular adhesion molecule-1 and vascular endothelial cadherin. The fibrin clot is stabilized by the transglutaminase factor XIIIa,153–155 whose function is to cross-link fibrin and other adhesive proteins, including integrin receptors, to provide a stable network (Fig. 34-10). The protransglutaminase factor XIII circulates in plasma as a tetramer and is also present as a dimer in platelets. Activation of the protein by thrombin cleavage and Ca2+ binding yields factor XIIIa, which cross-links fibrin by forming exopeptide bonds between the ε-amino lysyl residues and the γ-carboxamide groups of glutamine residues. The clot resulting from thrombin cleavage of fibrinogen to form the clot fibrin and factor XIII cross-linking create an insoluble network that, with platelets, seals the perforation in the blood vessel with a temporary scaffold, stopping further blood loss. This scaffold is ultimately removed in the fibrinolytic process and replaced by cellular reconstruction of the perforated vessel.
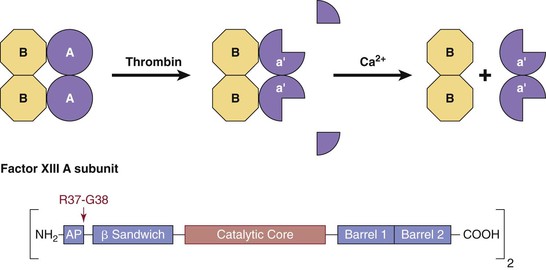
Figure 34-10 Plasma factor XIII. Thrombin-catalyzed activation of plasma factor XIII (A2B2; 320,000 Da) occurs in two steps. First, thrombin cleaves the R37-G38 bond. This releases the activation peptides (residues 1 to 37) from the A chains, producing the inactive intermediate a′2B2. In the second Ca2+-dependent step, the B chains dissociate from the a′2B2 intermediate, exposing the active site cysteine, Cys314, of the a′ subunits. The enzyme a′2 catalyzes the formation of isopeptide bonds between glutamine residues and lysine residues of adjoining polypeptide chains.
Fibrinolysis Proteins
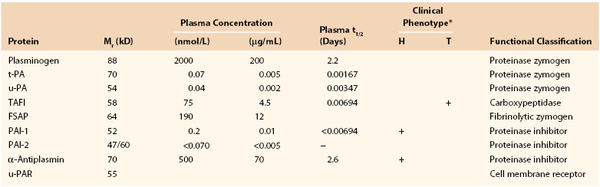
Clot formation is integrated with clot dissolution. The mechanisms of clot dissolution center on fibrin-specific reactions. The key proteins involved are plasminogen; the plasminogen activators tissue plasminogen activator (t-PA) and urokinase or urinary plasminogen activator (u-PA); thrombin; and the inhibitors PAI-1, α2-antiplasmin, and TAFIa (Fig. 34-11 and Table 34-3).
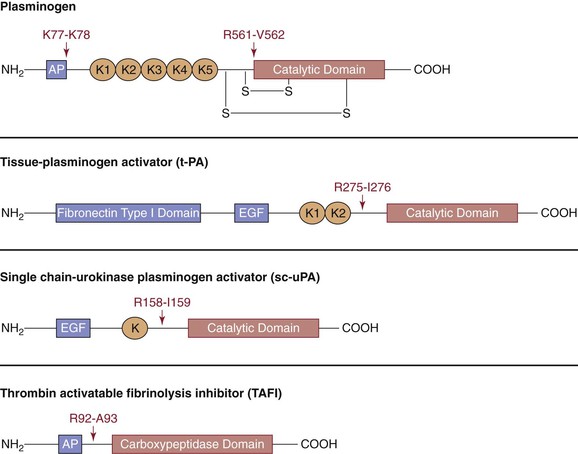
Figure 34-11 A schematic representation of proteins of the fibrinolytic system. Plasminogen, tissue-type plasminogen activator, urokinase plasminogen activator, and thrombin-activatable fibrinolysis inhibitor are shown with their various domains depicted. Cleavage sites with the specific amino acid residues are shown. AP, Activation peptide; K, kringle domain; EGF, epidermal growth factor domain.
Fibrinolysis Activators
Plasminogen/Plasmin.
Plasminogen is the inactive precursor of the enzyme plasmin, the primary catalyst of fibrin degradation.156 Hereditary plasminogen deficiency is described either as a deficiency of plasminogen antigen and activity (type I) or as a normal antigen level but reduced activity (type II, dysplasminogenemia).157 Plasminogen is also an acute phase reactant protein.158,159 Homozygous plasminogen-deficient mice are viable but exhibit severe thrombosis with systemic fibrin deposition and die prematurely.160 Plasminogen is synthesized in the liver and is present in a wide variety of tissues and body fluids, including saliva, lacrimal gland secretions, seminal vesicle fluid, and prostate secretions. The zymogen is converted to the serine protease plasmin by cleavage of the Arg561-Val562 peptide bond by the activators. Activation of plasminogen occurs through three pathways: (1) the intrinsic activator system (analogous to the contact system of blood coagulation), (2) the extrinsic activators (t-PA and u-PA), and (3) the exogenous activation system involving pharmacologic agents (thrombolytic drugs). The ordinary path in vivo appears to be the extrinsic pathway, catalyzed by t-PA and u-PA. The extent to which the intrinsic pathway functions is the subject of debate.
Plasminogen activation is primarily inhibited by PAI-1, which targets u-PA and t-PA. PAI-1 also has a role in tissue remodeling by interfering with the vitronectin-dependent processes of cell adhesion and migration.160,161 Congenital deficiency of PAI-1 is rare, with homozygous individuals displaying abnormal bleeding in response to trauma.161
In plasma or bound to fibrin in a blood clot, activation of plasminogen by either t-PA or u-PA converts Glu-plasminogen to Glu-plasmin. The two-chain enzyme can degrade fibrin, fibrinogen, and many other molecules. Glu-plasmin autodigests by cleaving itself at Lys77 to generate Lys-plasmin.162,163 Both Glu and Lys plasmin cleave after basic amino acid residues. The lysine analogues ε-aminocaproic acid and tranexamic acid can compete with lysyl residues in proteins for binding to plasminogen and hence are inhibitors of fibrinolysis.164,165
Inhibition of plasmin by α2-antiplasmin is the primary route for regulation of plasmin’s hemostatic function; suppression of plasmin activity beyond the locale of fibrin deposition is imperative if systemic fibrinogenolysis is to be prevented. Plasmin bound through its lysine binding sites to fibrin reacts more slowly with α2-antiplasmin than when it is free in solution. This differential reactivity effectively localizes plasmin activity to the fibrin clot.
Tissue Plasminogen Activator.
t-PA is predominantly a product of endothelial cells,166–170 but it is also produced by vascular smooth muscle cells,171 neuronal cells,172 megakaryocytes,173,174 mast cells,175,176 monocytes,177 and fibroblasts.178 Factors that regulate its secretion and release from the endothelium are important mediators of blood clotting or inflammation. These include thrombin, histamine, acetylcholine, bradykinin, epinephrine, interleukins, shear stress, and vaso-occlusion.179,180 Functional t-PA concentrations have been reported to be less than 20 pmol/L, with most t-PA found in complex with PAI-1.181–183 (The half-life in plasma is short; pharmacokinetic modeling indicates a half-life of 2.4 minutes for active t-PA and 5 minutes for the t-PA/PAI-1 complex.184,185) No cases of congenital deficiency of t-PA have been reported. Transgenic mice lacking a functional t-PA gene develop normally and display a normal basal hemostatic phenotype.186 Mice in which both the t-PA and u-PA genes are disabled have shortened life spans and experience severe spontaneous thrombotic episodes.186,187
Regulation of t-PA activity in blood is accomplished by three primary mechanisms: (1) control of its catalytic potential by the fibrin dependence of plasminogen activation; (2) control of systemic levels of functional t-PA by the concerted processes of rapid t-PA removal by hepatic clearance and inhibition by the circulating serpin PAI-1; and (3) control of t-PA activity levels at the site of injury by the competing processes of increased t-PA secretion by traumatized and recruited cells versus PAI-1 release by activated platelets.
t-PA manifests its full fibrinolytic potential only when it is bound to fibrin.188–190 This binding interaction aligns t-PA and plasminogen on the fibrin surface so that the catalytic efficiency of t-PA is enhanced several hundredfold. This is vital to the localization of plasmin generation at the site of fibrin deposition. Release of t-PA from the vessel wall is another important regulator of fibrinolysis.191,192 The rate at which clots lyse depends on how rapidly t-PA is secreted by the relevant cells in the vicinity of an injury.193,194 For example, activated platelets secrete serotonin, which can induce endothelial cells to release t-PA; they also release PAI-1 from their alpha granules. Although only a fraction of this PAI-1 is in the active form, it functions to downregulate plasminogen activation.195,196
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


