Chapter 37
Coagulopathy and Hemorrhage
Jeffrey H. Lawson, Kyla M. Bennett
Understanding the complex biology of coagulopathy and hemorrhage is imperative in the perioperative management of surgical patients. “Coagulopathy” is a term employed loosely in the literature and in the clinical setting. It is applied to patients without identifiable coagulation abnormality but with clinical bleeding or recurrent thrombosis as often as to patients with disseminated intravascular coagulopathy and significantly abnormal coagulation parameters or those with specific inherited factor deficiencies. The Gale Encyclopedia of Medicine defines coagulopathy as a disorder in which blood is either too quick or too slow to coagulate [clot].1 Coagulation is a physiologic defense mechanism aimed at maintaining the integrity of the circulatory system in the setting of vascular injury. There is a critical balance between coagulation and fibrinolysis aimed at preventing pathologic hemorrhage and thrombosis. The surgeon is thus tasked with preventing adverse events by achieving adequate hemostasis without effecting extensive thrombosis. This challenge is particularly germane to vascular surgery. An understanding of normal hemostasis, as outlined in Chapter 34, is necessary to assimilate the pathophysiology of the coagulopathies and treatments presented here. In this chapter, we review the pathophysiology, diagnosis, and treatment of perioperative hemorrhage.
Diagnosis and Preoperative Screening for Bleeding Disorders
Identifying patients preoperatively who are at risk for bleeding allows the surgeon to initiate corrective treatment before performing a surgical procedure and to plan for optimal perioperative management.
History and Examination
The key to minimizing perioperative hemorrhage is to identify patients at significant risk for hemorrhage or coagulopathy by means of a thorough history and physical examination. A detailed history should elicit any previous episodes of bleeding with prior surgery, dental extractions, or trauma as well as any easy bruising or epistaxis. A patient with a family history of significant hemorrhagic complications should be evaluated for increased bleeding risk. Complete drug and medication histories (including homeopathic or herbal remedies) are important to evaluate for agents that affect the clotting cascade. A history suggestive of hematuria or melena requires further investigation. Physical examination should identify any evidence of petechiae or excessive bruising.
Laboratory Testing
With the abundance of laboratory tests available for the evaluation of hemostasis, performing all of them is neither cost-effective nor necessary. One must adopt a systematic approach to selecting which, if any, laboratory studies a particular patient requires.
Risk Stratification
Rapaport devised the following four-level stratification scheme to determine the need for preoperative laboratory testing according to the patient’s clinical status and bleeding history, and the planned operation2:
Level I.
Patients classified as having level I risk have no bleeding history and will undergo minor procedures such as lipoma excision. For them, no further hematologic evaluation is required because the cost of testing outweighs the probability of finding an abnormality and the risk of bleeding, even if one exists.
Level II.
Level II risk patients have no previous bleeding history but will undergo a major operation. Normal activated partial thromboplastin time (aPTT) and platelet count should effectively eliminate the risk of life-threatening bleeding.
Level III.
Patients whose bleeding history raises concern for defective hemostasis and those whose procedure might impair hemostasis (e.g., cardiac or neurosurgical interventions) are classified as having level III risk. In these patients, preoperative evaluation of the following factors is appropriate:
1. Adequacy of hemostatic plug formation—bleeding time and platelet count
2. Adequacy of coagulation reactions—prothrombin time (PT) and aPTT
3. Size and stability of the fibrin clot—factor XIII deficiency and fibrinolysis screening
Level IV.
Patients are classified as having level IV risk if they offer history or have physical findings highly suggestive of abnormal hemostasis, and the surgical procedure is not a factor. In addition to performing the tests indicated for level III patients, the following should be considered (especially if the results of level III testing are normal):
1. Bleeding time after the administration of 600 mg of aspirin—to uncover von Willebrand disease (vWD) or a qualitative platelet disorder3
2. Factor VIII and factor IX levels
Proper Blood Drawing
A properly drawn blood sample is paramount to interpreting the results of clotting tests. Whole blood is collected into an evacuated sample tube containing a fixed amount of citrate as an anticoagulant. The ratio of whole blood to citrate solution should be 9 : 1. An adult sample tube should be filled to at least 60% to 80% of its full collection volume to avoid excessive anticoagulation (a pediatric sample tube needs to be filled to 90% of its volume).4 The anticoagulated blood should be mixed gently by being inverted three to four times. Testing should be done within 2 hours if the sample is kept at room temperature and within 4 hours if kept at 4° C. Table 37-1 summarizes the screening tests used to determine the cause of coagulopathy.
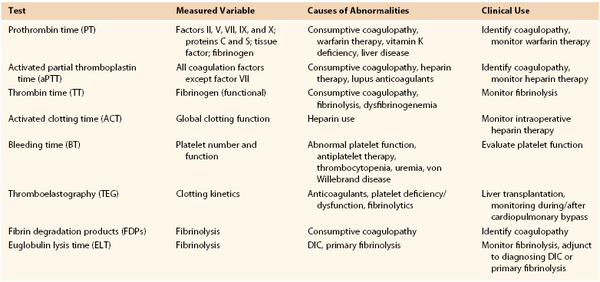
Specific Laboratory Tests
The specifics of the pathways and assays pertinent to prothrombin time, activated partial thromboplastin time, thrombin time, activated clotting time, and thromboelastography are well described in Chapter 34 (in the section “Blood Coagulation Monitoring”). Thus they are described only briefly here.
Prothrombin Time.
PT is used to assess the extrinsic pathway of clotting (factors II, V, VII, IX, and X and fibrinogen). The end point is the time required (in seconds) for a fibrin clot to form. Argatroban therapy and very high doses of heparin can also cause an elevated PT value secondary to thrombin (IIa) inhibition.5
Activated Partial Thromboplastin Time.
The aPTT is used to assess the intrinsic pathway (factors VIII, IX, XI, and XII) and final common pathway (factors II, V, and X and fibrinogen) of clotting. Primary uses of aPTT include monitoring heparin therapy and detecting lupus anticoagulant, hemophilia A, and hemophilia B.
Thrombin Time.
TT measures the conversion of fibrinogen to fibrin, which is the final step in the clotting pathway. Prolongation of TT is attributable to direct thrombin inhibitor (DTI) therapy, heparin, lytic administration, and dysfibrinogenemia.
Activated Blood Clotting Time.
The activated clotting time (ACT) test is performed by addition of a coagulant-accelerating matrix (e.g., diatomaceous earth) to a sample of whole blood and measurement of the time required for clot formation. This method is generally used to monitor adequacy of heparinization in the operating room or interventional suite.
Bleeding Time.
The bleeding time, or the time needed for a superficial wound to clot, is used to assess primary hemostasis. One measures bleeding time by making a controlled wound in the forearm or earlobe and subsequently measuring the time for clotting to occur.6 Clotting that takes longer than 5 minutes is considered abnormal. Bleeding time is mainly affected by the number and function of platelets, although vasoconstriction or vasodilatation may affect the bleeding time as well. Other factors that prolong the bleeding time include heparin therapy (as a result of platelet inhibition), vWD, thrombocytopenia, aspirin or other antiplatelet therapy, and uremia.
Thromboelastography.
Thromboelastography (TEG) measures real-time clotting performance and in situ fibrinolysis. It is used for intraoperative monitoring of anticoagulation (e.g., cardiac surgery) and monitoring of fibrinolytic administration.
Fibrin Degradation Products.
Fibrin degradation products (FDPs) occur as the result of primary or secondary fibrinolysis. Elevations of their levels are seen in disseminated intravascular coagulation (DIC), cirrhosis, eclampsia, and trauma and following lytic therapy. The FDP assay has few specific clinical applications except in the attempt to identify primary fibrinolysis.
Euglobulin Lysis Time.
The euglobulin lysis time (ELT) is an assay used to assess global function of the plasma fibrinolytic system. It measures the time required for a clot to lyse in a test tube (the normal value is 90 to 240 minutes). For this test to be useful, a patient must have adequate clot formation. This test is used as part of a panel to help differentiate a primary fibrinolytic state from DIC. A shortened ELT in patients without thrombocytopenia or schistocytosis is diagnostic of primary fibrinolysis, whereas a shortened ELT with thrombocytopenia or schistocytosis probably indicates DIC. Reports imply that ELT can also be used to determine the presence of excessive tissue plasminogen activator t-PA and deficiency of plasminogen activator inhibitor.7 A drawback of this assay is its significant reported interlaboratory variability.8
Inherited Coagulopathies
Platelet Disorders
Inherited platelet disorders are rare conditions with varying degrees of phenotypic severity. These disorders affect multiple aspects of platelet function, namely aggregation, secretion, adhesion, and procoagulant activity. There are a large number of rare conditions, the most common of which are described in the following sections.
von Willebrand Disease
von Willebrand factor (vWF) plays an important role in primary hemostasis by binding to both platelets and endothelial components, effecting formation of an adhesive bridge between them. vWF also contributes to clot formation by acting as a carrier protein for factor VIII.9
von Willebrand disease is the most common inherited bleeding disorder. Although it affects about 1% of the population, only 5% of affected patients are symptomatic.10 Clinical manifestations range from nil to severe bleeding, depending on the level of functional, circulating vWF. Easily identifiable but nonspecific symptoms include easy bruising, mucous membrane bleeding, prolonged epistaxis, postoperative bleeding, gastrointestinal bleeding, and heavy menstrual bleeding.11,12 Previously undiagnosed patients sometimes have the first manifestation of the disease after taking antiplatelet agents such as nonsteroidal anti-inflammatory drugs (NSAIDs) and aspirin.
vWD is classified into three major types on the basis of clinical laboratory test results and genetic mutations, as delineated in Table 37-2.
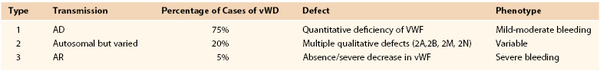
Type 2 comprises at least four subtypes with variable inheritance patterns. Patients with type 2A vWD (≈15% of all with vWD) have decreased platelet-dependent functions owing to loss of high-molecular-weight multimers. Those with type 2B disease (≈5% of all with vWD) have vWF with increased affinity for platelet glycoprotein Ib (GP-Ib). Type 2M is less common and results in reduced binding of vWF to platelet GP-Ib despite the presence of large vWF multimers. Patients with type 2N disease have mutations that alter the vWF binding site for factor VIII. Impaired binding effects rapid clearance of factor VIII.10
If vWD is suspected, the initial screening tests are: (1) bleeding time, (2) closure time (vWF-dependent platelet function), (3) platelet count, and (4) aPTT. If results of these studies are positive or suspicious, more specific testing is pursued (Table 37-3).9,13–15
Table 37-3
Tests for von Willebrand Disease
| Test | Evaluates | Type in Which Abnormality Seen |
| Plasma vWF antigen | Quantitative defects (presence) | 1,3 |
| Ristocetin cofactor activity | Agglutination requires largest vWF multimers | 2A, 2M |
| Factor VIII coagulant activity | Factor VIII half-life and secretion depend on vWF | 1,3 |
| Ristocetin-induced platelet aggregation (RIPA) | vWF concentration and affinity for GP-Ib | 2A, 2M, 3 |
| vWF multimer distribution | Presence of high-molecular-weight multimers | 2A, 2B |
| vWF–factor VIII binding assays | Affinity of factor VIII–vWF binding | 2N (versus hemophilia A) |
GP, Glycoprotein; vWF, von Willebrand factor.
Treatment decisions are based on severity of symptoms and type of vWD. Symptomatic patients should avoid nonsteroidal anti-inflammatory drugs. Desmopressin (DDAVP) is effective in patients with type I vWD perioperatively and in those with mild to moderate bleeding episodes. Patients with type 3 and severe forms of type 2A, 2B, and 2M disease usually require replacement therapy with vWF, factor VIII–vWF concentrates, or cryoprecipitate. In general, the goal of treatment in these patients is to maintain the activity of factor VIII and vWF between 50% and 100% for 3 to 10 days to address episodes of serious bleeding or for major surgery.11
Giant Platelet Disorders
Giant platelet disorders are a group of rare disorders characterized by thrombocytopenia, large platelets, and variable bleeding symptoms. They are generally subcategorized into four groups: those with a structural defect (e.g., Bernard-Soulier syndrome with glycoprotein abnormalities), those with abnormal neutrophil inclusions (e.g., May-Hegglin anomaly), those with systemic manifestations (e.g., hereditary macrothrombocytopenia with hearing loss), and those with no specific abnormalities (e.g., Mediterranean macrothrombocytopenia).16
Bernard-Soulier syndrome, the most common of these platelet disorders, is characterized by thrombocytopenia, large platelets, and bleeding. It is attributed to dysfunction or absence of the GP-Ib/IX/V complex, which is a primary adhesion receptor of platelets.16 The disorder manifests early in life as bleeding symptoms, most frequently epistaxis or gingival or cutaneous bleeding. Frequently, a severe hemorrhagic episode is noted after surgery (e.g., circumcision). Laboratory findings include thrombocytopenia ranging from less than 30 to 200 × 103/µL (normal) and a prolonged bleeding time with normal clot retraction.16,17 Management of patients with this syndrome usually entails education and avoidance of minor trauma. In the event of significant hemorrhage, platelet transfusion is generally indicated.18
Glanzmann’s Thrombasthenia
Glanzmann’s thrombasthenia is an autosomal recessive disease with a large number of reported mutations. A defect in GP-IIb/IIIa renders platelets unable to aggregate.19,20 Normal GP-IIb/IIIa allows platelets to bind soluble proteins and vWF. In Glanzmann’s thrombasthenia, platelets can attach to exposed endothelium but cannot form aggregates. A wide spectrum of phenotypic severity is reported, but mucocutaneous bleeding and absence of platelet aggregation are classic findings.21 Significant bleeding episodes typically require platelet transfusion.16,19,22
Storage Pool Disorders
Storage pool disorders result from platelet granule deficiencies. The granules are usually divided into two groups: alpha granules (which contain vWF, thrombospondin, fibrinogen, and platelet-derived growth factor) and dense granules (which release adenosine diphosphate [ADP] and serotonin). Gray platelet syndrome is an example of an alpha-granule storage disorder.23 This autosomal recessive disorder results in granules deficient in secretory proteins. Manifestations are typically limited to mucosal bleeding, but trauma-associated hemorrhage can occur. Laboratory analysis reveals moderate thrombocytopenia and a prolonged bleeding time. Preprocedural DDAVP and platelet transfusions form the basis for treatment.
Dense-granule deficiencies include Chédiak-Higashi syndrome, Wiskott-Aldrich syndrome, and thrombocytopenia–absent radius syndrome.24 Wiskott-Aldrich syndrome is an X chromosome–linked immunodeficiency manifested as thrombocytopenia with platelets of reduced size and function as well as eczema.25 A defect in glycoprotein L115 (a leukocyte/platelet surface molecule) yields platelets that are unable to form aggregates.25,26 Patients with the syndrome are typically diagnosed in early childhood with the constellation of thrombocytopenia, atopic dermatitis, and frequent infections. The only curative therapy is bone marrow transplantation. If platelet transfusion is required prior to transplantation, HLA-selected platelets should be used and all blood products should be irradiated.26
Hemophilia
The hemophilias are inherited bleeding disorders caused by low concentrations of specific coagulation factors.
Pathogenesis
The most common hemophilias are X-linked deficiencies of factor VIII (hemophilia A) and factor IX (hemophilia B).27 Factor XI deficiency (hemophilia C) is a less common autosomally transmitted disorder. Factor VIII is a complex plasma glycoprotein produced by liver sinusoidal endothelial cells and by vascular endothelial cells.28 It has a half-life of 12 hours in adults and is protected from premature degradation by vWF. Factor IX is a vitamin K–dependent protein synthesized by the liver with a plasma concentration approximately 50 times that of factor VIII and a half-life of 24 hours.29 Bleeding in hemophilia results from a failure of secondary hemostasis. Although a normal platelet plug forms, stabilization of the plug by fibrin is defective owing to inadequate amounts of thrombin.
Diagnosis
Hemophilias A and B are clinically indistinguishable. A diagnosis of hemophilia is confirmed by an assay for the specific factor. However, factor VIII deficiency secondary to hemophilia A requires distinction from vWD by assays for vWF. Depending on the concentration of the factor, the bleeding tendency is classified as mild, moderate, or severe.30 Mild disease has a factor VIIIc or IXc concentration between 0.05 and 0.40 IU/mL (5%-40% of normal) with bleeding manifestations after surgery, dental intervention, or trauma. Spontaneous bleeding is not seen. Moderate disease translates to a factor VIIIc or IXc concentration of 0.01 to 0.05 IU/mL (1% t-5% of normal). This concentration can result in bleeding into muscles and joints following minor injury. Major surgical or dental intervention effects severe hemorrhage.29 Severe disease is seen with a concentration of factor VIIIc or IXc of less than 0.01 IU/mL (<1% of normal).
Though most cases of hemophilia are identified via family history, some appear as de novo mutations. Whereas severe disease is typically diagnosed by 2 years of age, mild or moderate disease may not be recognized until adulthood. The hallmark of severe hemophilia is spontaneous bleeding into joints and muscles.
Management
The primary goal of treatment is to sufficiently increase the concentration of the missing factor to stop or prevent spontaneous, traumatic, or surgical hemorrhage. A critical aim is to aggressively treat and prevent joint bleeding, which otherwise can lead to crippling arthropathy and joint deformity.31 Primary prophylaxis (prior to any episode of joint hemorrhage) to prevent this devastating complication is now recommended, with most centers recommending initiation prior to 3 years of age.
Although DDAVP can sufficiently raise factor VIII levels in patients with mild hemophilia A, factor concentrations are typically required for more significant disease. Multiple factor VIII and IX concentrations are available as either recombinant or plasma-derived products. For prophylaxis of a nonbleeding patient, a factor VIII concentration of 3% is considered adequate. A level of 30% is required for minor bleeding, and 50% for major bleeding. Prior to elective surgery, a factor VIII level greater than 80% is recommended, with a goal of 30% to 50% to be maintained in the first two postoperative weeks. The goals for hemophilia B include a factor IX level of 30% for minor bleeding and greater than 50% for major bleeding.32
In some patients who undergo long-term replacement therapy, antibodies against factors VIII and IX develop. For these patients, recombinant factor VIIa (rFVIIa) or activated prothrombin complex concentrates might be necessary to achieve hemostasis. These agents act by bypassing the need for factors VIII and IX to achieve coagulation. The long-term goal for patients with high antibody titers is immune tolerance induction therapy, which results in a gradual reduction of antibody titers and increased tolerance of factor replacement therapy.
Acquired Coagulopathies
Platelet Disorders
Acquired platelet dysfunction has myriad causes, including uremia, liver dysfunction, cardiopulmonary bypass, hypersplenism, hematologic malignancy, thrombotic thrombocytopenic purpura, and immune thrombocytopenic purpura. These processes can result in quantitative or qualitative platelet deficiencies and should be considered in any investigation of abnormal bleeding, especially that accompanied by thrombocytopenia.
Liver disease evokes thrombocytopenia via portal hypertension with splenic sequestration as well as by decreased thrombopoietin production. Cardiopulmonary bypass and mechanical assist devices place platelets in contact with nonphysiologic machine surfaces, which result in pathologic alterations in surface glycoprotein expression. Activation of these glycoproteins results in granule release as well as decreased endothelial adhesion and aggregate formation.
Uremia
Although bleeding diatheses secondary to uremia are not completely understood, it is clear that platelet dysfunction plays a critical role. Uremia results in both intrinsic platelet defects and alteration of platelet-endothelial interaction.33 Changes have been described in GP-IIb/IIIa, in ADP release from alpha granules, and in the arachidonic acid pathway.34
Uremic patients can exhibit easy bruising and mucosal bleeding. Laboratory evaluations demonstrate a prolonged bleeding time in the setting of normal values for PT, aPTT, and platelet count. Directed treatment is typically reserved for major surgery or active bleeding. In this setting, general recommendations include correcting anemia to a hematocrit of 25% to 30% (to improve platelet aggregation and adhesion) and administration of DDAVP (to enhance release of vWF) and/or conjugated estrogen (unknown mechanism).35 Dialysis is the most effective treatment for uremic complications.
Vitamin K–Related Disorders
Factors II, VII, IX, and X as well as proteins C and S require vitamin K for activation. Functional impairment of these factors is due to failure of vitamin K–dependent carboxylation. The cause is typically lack of vitamin K (malnutrition or malabsorption due to biliary obstruction or gut bacterial overgrowth) or pharmacologic blockade of vitamin K (warfarin).
Treatment entails either functional factor replacement (FFP) or vitamin K (allowing carboxylation of patient’s factors) to address active bleeding or in anticipation of surgical intervention. FFP acts quickly as a direct replacement without need for enzymatic reactions; it has an immediate effect that lasts about 6 hours. Vitamin K takes at least 6 hours to take effect but exerts a more durable reversal.
Disseminated Intravascular Coagulation
DIC is systemic activation of the coagulation cascade that induces both thrombosis and hemorrhage.
Pathogenesis
DIC is primarily caused by uncontrolled production of thrombin, which leads to systemic intravascular deposition of fibrin. The inciting factor is generally vascular injury or intravascular exposure to a procoagulant, which results in initiation of normal hemostasis. Normally, thrombin generation is tightly regulated by a combination of intravascular and endothelial factors. In DIC, these inhibitory mechanisms become dysfunctional or overwhelmed, leading to the production of excess thrombin. The increased circulating levels of thrombin provoke greater fibrin production with microvascular deposition. Thrombosis of the microvasculature causes the end-organ damage seen in severe DIC. The systemic thrombosis activates tissue plasminogen activator, which causes thrombolysis, consumptive coagulopathy, and bleeding diathesis.
Etiology
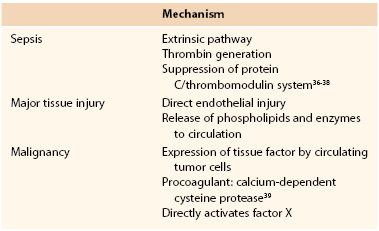
The most common precipitants of DIC are sepsis, extensive traumatic tissue injury, cancer, amniotic fluid embolism, and placental abruption. The final common pathway for all of these entities is systemic activation of the coagulation cascade. Proposed specific mechanisms are delineated in Table 37-4.36–39
Clinical Phases
Even though DIC is a continuously progressing cascade of events, it can be described as three major clinical phases.40 They are described here, and their typical laboratory findings are delineated in Table 37-5.
Table 37-5
Coagulation Parameters in Disseminated Intravascular Coagulation
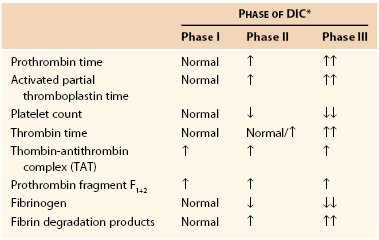
* Phases of DIC are defined as follows:
DIC, Disseminated intravascular coagulation; ↑, increased (no. of arrows indicates extent of increase); ↓, decreased (no. of arrows indicates extent of decrease).
Management
The overriding goal in the management of DIC is correction of the precipitating disease. Supportive therapy generally necessitates replacement of clotting factors and platelets while the inciting process is addressed. In patients with active bleeding, very aggressive replacement is pursued. Goals of resuscitation include a fibrinogen level over 100 mg/dL. Management also requires achieving and maintaining normothermia and adequate tissue perfusion. There is little evidence to support heparin therapy in acute DIC. During the supportive phase, emphasis should be put on correcting the underlying pathologic process, the achievement of which will, in turn, stop the activation of the cascade. For instance, in septic patients, broad-spectrum antibiotics, abscess drainage, and débridement of infected or necrotic tissues is critical.
Primary Fibrinolysis
Fibrinolytic system derangements can result in either excessive bleeding or thrombosis. It is essential to differentiate primary fibrinolysis from secondary fibrinolysis (DIC) because of their differing management strategies.
Fibrinolytic System
For an essential complete review of the fibrinolytic system, please refer to Chapter 34. In brief: Plasminogen circulates as an inactive zymogen. Activation of plasminogen by t-PA or urinary-type plasminogen activator (u-PA) results in its conversion to plasmin, a serine protease that degrades fibrin. Inhibitors of plasmin include plasminogen activator inhibitor (PAI-1; inhibits tissue and urinary-type plasminogen activators) and α2-antiplasmin (hepatic production). Thrombin-activatable fibrinolysis inhibitor (TAFI; a procarboxypeptidase activated by thombin-antithrombin complex [TAT]) also constrains fibrinolysis by cleaving the C-terminal lysine residue of fibrin fragments. This disruption prevents recruitment of plasminogen to a partially digested clot, thereby inhibiting fibrinolysis.
Pathogenesis
In this complex plasmin-plasminogen activator system, excessive bleeding can result from hyperactivity of plasminogen activators or deficient activity of fibrinolysis inhibitors. In primary fibrinolysis, there is pathologic direct activation of plasminogen. Excess circulating plasmin saturates plasminogen activator inhibitor-1 and α2-antiplasmin and cleaves fibrin and clotting factors, including factors V and VIII. Plasmin generated on the surfaces of platelets and endothelium inappropriately lyses hemostatic plugs. To counter the excess plasmin and fibrinolysis, excess thrombin is generated, resulting in microvascular thrombosis.
Etiology/Diagnosis
A primary hyperfibrinolytic state can develop in multiple clinical settings, including malignancy, liver failure, trauma, and congenital deficiency of fibrinolysis inhibitors.41,42 Differentiating primary fibrinolysis from a secondary process such as DIC can be exceedingly difficult. The simplest laboratory findings to diagnose primary fibrinolysis are a short euglobulin lysis time in the absence of thrombocytopenia and the presence of schistocytes. Though challenging, the distinction between these two entities is critical as they require quite dissimilar treatments.
Management
The crux of treatment is inhibition of fibrinolysis using lysine analogues. The lysine analogues, aminocaproic acid and tranexamic acid, act as competitive inhibitors of the conversion of plasminogen to plasmin.43 Often, factor replacement is also required, typically factors V and VIII. Prior to its removal from the market, aprotinin was also used as a therapy for this disorder.44
Drug-Induced Coagulopathies
Heparins
Heparin is a naturally occurring, highly sulfated glycosaminoglycan. Heparin prevents clot formation and propagation by binding to and activating antithrombin. The resulting conformational change in antithrombin accelerates its ability to inactivate factors II (thrombin), Xa, and IXa.45 Heparin can be monitored via ACT or aPTT, both of which demonstrate prolongation. Heparin is available for subcutaneous (half-life 8 hours) or intravenous (half-life 1.5 hours) administration. Intravenous heparin is frequently used for intraprocedural anticoagulation because of its short half-life, point-of-care monitoring, and ease of reversal (protamine at 1 mg per 100 U intravenous heparin).46
Enzymatic depolymerization of unfractionated heparin results in fragments that are approximately a third the size of heparin. These are marketed as low-molecular-weight heparins (LMWHs; such as enoxaparin and fondaparinux). Administered subcutaneously, these agents offer a longer half-life and greater bioavailability than heparin.46 Benefits of LMWHs include ability to be given once or twice daily and the lack of need for laboratory monitoring for therapeutic use. The risk of bleeding with LMWH prophylaxis is low and comparable to that seen with prophylactic subcutaneous heparin. Withholding fully therapeutic LMWH for 24 hours prior to major surgical intervention is recommended. There is no described benefit in protamine for reversal of LMWHs.47
As delineated further elsewhere (see Chapters 35 and 38), use of either type of heparin risks heparin-induced thrombocytopenia (HIT). Briefly, patients with this disorder form antibodies to the complex of heparin and platelet factor 4 on platelet surfaces. The presence of the antibodies leads to uncontrolled platelet activation and pathologic thrombosis.
Warfarin
Warfarin is an orally administered vitamin K antagonist (VKA). It acts by preventing vitamin K–dependent carboxylation of factors II, VII, IX, and X as well as of proteins C and S. The liver synthesizes nonfunctional forms of these proteins, which require carboxylation to become active. Following initiation, warfarin typically requires 2 or 3 days to take effect. Owing to the relatively shorter half-lives of proteins C and S in comparison with the clotting factors, a heparin bridge is sometimes required at the initiation of warfarin therapy to prevent sequelae of a paradoxically hypercoagulable state. The earlier loss of the anticoagulant effect of protein C or S (more commonly problematic in patients with protein C or S deficiency) can result in small vessel thrombosis, which manifests as warfarin-induced skin necrosis. This entity, if encountered, is treated with cessation of warfarin and initiation of therapeutic heparin.48
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


