Noninfectious Pulmonary Complications of Hematopoietic Stem Cell and Solid Organ Transplantation
HEMATOPOIETIC STEM CELL TRANSPLANTATION
Following a brief overview of the topic, we present a discussion on the frequency of noninfectious complications, associated risk factors, and the impact of noninfectious complications on outcome.
 OVERVIEW
OVERVIEW
Important considerations in hematopoietic stem cell transplantation are discussed below. Subsequently, solid organ transplantation is discussed in a separate section.
Hematopoietic stem cell transplantation (HSCT) primarily is used to treat hematological and lymphoid cancers, selected solid tumors, and nonneoplastic diseases including autoimmune disorders, amyloidosis, and aplastic anemia.1 Over 30,000 autologous and 25,000 allogeneic HSCTs are performed annually worldwide.2 The most common graft source is peripheral blood.2 Other graft sources include bone marrow and cord blood. A total of 7892 allogeneic and 12047 autologous HSCTs were performed in 2011 in the United States.2 The main indications for autologous transplant include multiple myeloma and lymphomas, and allogeneic transplant is most commonly performed for acute and chronic leukemia, lymphoma, and myelodysplastic syndrome. A conditioning regimen is employed before transplantation to eradicate malignant cells and, in allogeneic transplantation, to induce immunosuppression that permits engraftment.1 Some patients are also given total-body irradiation for myeloablation and immunosuppression. The conditioning regimen can be termed myeloablative, reduced intensity, or nonmyeloablative.3
Following HSCT, the immune system recovers along predictable patterns depending on the underlying disorder, stem cell source, and complications such as graft versus host disease (GVHD).4 Recovery occurs faster in autologous recipients, in those who receive peripheral blood stem cell grafts, and after nonmyeloablative conditioning. The posttransplant period is divided into three phases: Pre-engraftment, early posttransplant, and late posttransplant.5 The pre-engraftment phase (0–30 days) is characterized by neutropenia and breaks in the mucocutaneous barriers. The early post-engraftment phase (30–100 days) is dominated by impaired cell-mediated immunity. The impact of this cell-mediated defect is determined by the development of GVHD and the corresponding immunosuppressant medications. The late posttransplant phase (>100 days) is characterized by defects in cell-mediated and humoral immunity, as well as function of the reticuloendothelial system in allogeneic transplant recipients. The development of noninfectious pulmonary complications follows characteristic temporal patterns.6 Pulmonary edema, diffuse alveolar hemorrhage (DAH), and peri-engraftment respiratory distress syndrome (PERDS) usually occur during the first 30 days posttransplant (Fig. 95-1). Idiopathic pneumonia syndrome (IPS) can occur at any time following transplant.
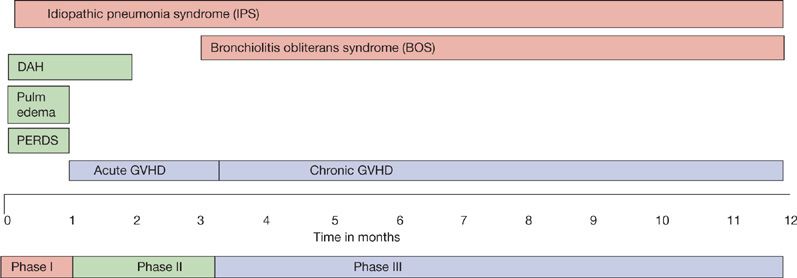
Figure 95-1 The temporal pattern of major noninfectious pulmonary complications following hematopoietic stem cell transplantation.
The mortality of patients following HSCT is high and depends on the underlying disease and type of transplant.2 Patients receiving human leukocyte antigen (HLA)-identical sibling transplants for acute myelogenous leukemia (AML) in remission have a 100-day mortality rate of 7% to 9%, compared with 22% for patients with active leukemia at the time of transplantation. Early mortality after an unrelated donor transplant is higher than after an HLA-identical sibling transplant, but the rate also depends on the disease and stage. The causes of death in the first 100 days posttransplant mainly relate to the primary disease, GVHD, infection, and end-organ damage. IPS accounts for 1% to 5% of deaths. After an autologous transplant, recurrence or progression of primary disease is the most commonly reported cause of death. Among allogeneic transplant recipients, unrelated donor transplants have fewer deaths related to the primary disease; however, deaths related to organ failure and infections are more frequent.
 FREQUENCY OF NONINFECTIOUS PULMONARY COMPLICATIONS
FREQUENCY OF NONINFECTIOUS PULMONARY COMPLICATIONS
Many pulmonary complications occur in HSCT recipients (Table 95-1). Earlier reports identified pulmonary complications in 30% to 60% of HSCT recipients.7–9 Recent publications suggest changes in the pattern and frequency. In a study of 70 patients, pulmonary complications developed in 18 (25.7%), of whom only 2 had noninfectious pulmonary complications, one cryptogenic organizing pneumonia (COP), and another IPS.10 In a study of T-cell–depleted HSCT recipients, pulmonary complications developed in 16.5%.11 In a more recent report of autologous HSCT recipients, pulmonary complications developed in 27.6%, with noninfectious diagnoses in 10.2%.12 The reported rates of respiratory failure in allogeneic HSCT recipients also have declined over time.13
In a group of pediatric HSCT recipients, pulmonary complications were found in 90 of 363 (25%).14 In a study of pediatric allogeneic HSCT recipients, late-onset noninfectious pulmonary complications developed in 10 of 97 (10.3%), including 8 COP and 2 IPS.15 In autopsy series, the rate of pulmonary complications exceeds 80%.16,17
 RISK FACTORS FOR NONINFECTIOUS PULMONARY COMPLICATIONS
RISK FACTORS FOR NONINFECTIOUS PULMONARY COMPLICATIONS
Several factors have been implicated as risk factors for pulmonary complication in HSCT recipients (Table 95-2).5,11,14,15,18–23 It is controversial whether impaired pulmonary function before stem cell transplant is a risk factor for the development of early posttransplant respiratory failure and mortality. Studies that have examined the predictive value of pretransplant pulmonary function tests suggest that poor lung function before transplant increases the risk for posttransplant pulmonary complications24–28 and mortality.25,29,30 These findings were not confirmed in a large study.31 In addition, these analyses disagreed on which pretransplant lung function parameters were the strongest predictors of pulmonary complications and mortality, and they were limited by relatively small cohorts. A retrospective analysis of 2852 HSCT recipients assessed the association of pretransplant FEV1, FVC, total lung capacity (TLC), diffusing capacity for carbon monoxide (DLCO), and the development of early respiratory failure and mortality.32 Patients who developed early respiratory failure were more likely to have impaired lung function prior to transplant. Univariate analysis demonstrated that pretransplant FEV1, FVC, TLC, and DLCO were more likely to be reduced among patients who developed early respiratory failure.32 In a recent report of 1243 autologous HSCT recipients, pretransplant DLCO and underlying disease were the only independent risk factors for the development of pulmonary complications.12 In a study of 307 autologous and allogeneic HSCT recipients, 170 developed venoocclusive disease of the liver, and this was associated with reduced pretransplant DLCO.33 Advanced stage of the underlying disease at transplant is associated with the development of late-onset noninfectious pulmonary complications.22 Nonmyeloablative conditioning is associated with reduced pulmonary complications.34 Also, the incidence of pulmonary complications is low after T-cell–depleted stem cell transplant.11 Due to the absence of GVHD and the infrequent use of immunosuppressant medications and radiation therapy, noninfectious complications are less common in the autologous HSCT recipient.
 IMPACT OF PULMONARY COMPLICATIONS ON PATIENT OUTCOME
IMPACT OF PULMONARY COMPLICATIONS ON PATIENT OUTCOME
The mortality rate of allogeneic HSCT recipients has declined over time.13 A 2006 report found median survival of 41 weeks for patients with pulmonary complications compared with 350 weeks for those without complications.11 In patients who survived beyond 2 years after transplant, pulmonary complications accounted for 5% of the subsequent deaths, and patients with pulmonary complications had a 15-fold increased risk of death.35 In a study of pediatric allogeneic HSCT recipients, the 5-year posttransplant survival of patients with noninfectious pulmonary complications was 28.0% compared with 87.2% of those without complications.15 In an autopsy study of HSCT recipients, pulmonary complications were the only cause of death in 64% and one of multiple causes in 10%.16 There is controversy in the provision of intensive care for severe pulmonary complications. Respiratory failure accounts for about one-third of ICU admissions following HSCT, and early reports observed nearly 100% short-term mortality for HSCT recipients requiring ventilator support. More recent series report improving outcomes, with overall hospital mortality approximately 50% after ICU admission, and mortality 75% to 85% following mechanical ventilation.36–38 Worse outcomes are associated with advanced underlying disease, GVHD, multiple organ failure, and the need for vasopressors. Intensive care and aggressive management are usually warranted in the acute situation, especially during the engraftment period, to assess for potentially reversible conditions.
 APPROACH TO THE DIAGNOSIS OF NONINFECTIOUS PULMONARY COMPLICATIONS
APPROACH TO THE DIAGNOSIS OF NONINFECTIOUS PULMONARY COMPLICATIONS
The diagnostic approach to HSCT recipient with suspected noninfectious pulmonary complications should follow a systematic approach.6 Recipients with respiratory symptoms and signs usually are evaluated with a chest radiograph followed by high-resolution computed tomography (HRCT). If the radiographic evaluation fails to reveal pulmonary infiltrates, especially in allogeneic recipients, PFT should be performed to assess for bronchiolitis obliterans syndrome (BOS). For patients with pulmonary infiltrates, noninvasive tests should be performed for infections and other causes. Noninvasive approaches include sputum for microbiology studies, blood culture, blood aspergillus assay, cytomegalovirus (CMV) viral load testing, and urine antigen for Legionella pneumophila and Pneumococcus pneumonia.39 If noninvasive testing is nondiagnostic, invasive techniques should be considered, balancing the risk–benefit ratio. The invasive approach usually relies on fiberoptic bronchoscopy with bronchoalveolar lavage (BAL) and occasionally transbronchial lung biopsy. Other invasive procedures such as image-guided transthoracic needle aspiration and video-assisted thoracoscopic biopsy rarely may be needed.40
 PULMONARY FUNCTION ABNORMALITIES FOLLOWING HSCT
PULMONARY FUNCTION ABNORMALITIES FOLLOWING HSCT
Although the value of pretransplant PFT in predicting posttransplant outcome is somewhat controversial, many institutions perform pretransplant PFT as a baseline reference on all recipients, and posttransplant testing for allogeneic recipients.26,41,42 Some patients develop PFT abnormalities in the absence of respiratory symptoms.43 Restrictive and obstructive ventilatory defects, and gas transfer abnormalities are frequent long-term sequelae following allogeneic transplantation. A systematic review of data regarding allogeneic HSCT recipients reported decreased DLCO in 83%, restriction in 35%, and obstruction in 23%.44 A more recent study of over 500 allogeneic HSCT recipients documented somewhat lower frequencies: Impaired DLCO in 35%, restriction in 12%, and obstruction in only 6% of long-term survivors.45 While the frequency of restrictive defect and of impaired DLCO appeared constant over time, this study suggested a declining frequency of airflow obstruction. HSCT recipients with IPS and COP present with restrictive pulmonary function impairment, and those with BOS demonstrate airflow obstruction.46 The development of any posttransplant PFT abnormality is associated with increased risk of death.24,25,45,47
Risk factors for the development of PFT abnormalities following HSCT include smoking history, pretransplant pulmonary infection, viral infection in the early posttransplant period, older age, underlying disease, pretransplant chemotherapy and conditioning regimen, GVHD, and HLA mismatch.29,48–50 Respiratory muscle weakness, an alternative cause of restrictive PFT abnormalities, was reported in 52% of allogeneic HSCT recipients in a retrospective study.51 Risk factors for respiratory muscle weakness include chemotherapy, total-body irradiation, high-dose corticosteroids, immobility, and GVHD.51
 UPPER AIRWAY COMPLICATIONS
UPPER AIRWAY COMPLICATIONS
Significant injury to the mucosal barrier occurs in about 75% of HSCT recipients.52 Total-body irradiation, allogeneic transplant, leukemia, and delayed neutrophil engraftment are risk factors for mucositis.53 Upper airway inflammation due to mucositis may lead to laryngeal edema, dysphagia, and aspiration pneumonia. Life-threatening upper airway complications are more common in children.54,55 More severe mucositis is associated with an increased risk of secondary infection, requirement for narcotics, longer duration of parenteral nutrition and hospital length of stay, and increased overall mortality.53,56 Upper airway injury in HSCT recipients is usually managed with supportive care, including local symptomatic therapy and endotracheal intubation in severe cases.
 BRONCHIOLITIS OBLITERANS
BRONCHIOLITIS OBLITERANS
Bronchiolitis obliterans (BO) is an inflammatory and fibroproliferative process primarily affecting the small airways that leads to the presence of airflow limitation.57 Although BO can be idiopathic, it is more often associated with connective tissue disease, inhaled toxins, infections, drugs, chronic rejection following lung transplantation, and chronic GVHD.57–59 GVHD is a frequent complication of allogeneic HSCT, and is commonly associated with lung disease.48,59–61 The pulmonary manifestations of GVHD include diffuse alveolar damage, lymphocytic bronchitis/bronchiolitis with interstitial pneumonitis, COP, and BO.62 BO has only rarely been reported in autologous HSCT recipients.63
Frequency
The rate of BO varies among studies, depending on patient population. The lack of precise definition and uniform diagnostic criteria contribute to these variations. Although some studies have included pathological findings, most of the reported cases of the syndrome in HSCT recipients were defined by the presence of airflow limitation in the appropriate clinical setting.48
In a previous review of nine studies including 2152 allogeneic HSCT recipients, BO was reported in 8.3%, with a range of 6% to 20% in long-term survivors.58 In seven more recent studies, including 5543 allogeneic HSCT recipients, the average BO rate was 3.7%, with range of 2.6% to 10.3%.11,22,60,61,64–66
Risk Factors
Several factors are implicated as risks for BO.58 The most common is GVHD.25,61,64,65,67,68 In one study of allogeneic HSCT recipients, 6% of those with chronic GVHD developed BO compared to none without GVHD.69 Data from the International Bone Marrow Registry of 6275 adults with leukemia who received HLA-matched sibling transplant from 1989 to 1997, and survived at least for 100 days, showed that BO was associated with busulfan-based conditioning, time from disease diagnosis to transplant of 14 months or longer, peripheral blood stem cell source, transplant from female donor to male recipient, acute GVHD, and interstitial pneumonia.67 Older recipient or donor age, methotrexate use, and serum immunoglobulin deficiency are also implicated to be risk factors for BO.25,61,69–72 While pulmonary infections are common in patients with BO, it is not clear whether they are causally related or result from associated immunodeficiency.
Pathogenesis
Little is known about the pathogenesis of BO. Early in the syndrome, bronchiolar inflammation has been identified.62,73 The histopathology begins with lymphocyte inflammation around small vessels and the respiratory epithelial lining of the small airways, followed by epithelial cell necrosis and mucosal denudation.68 Two different forms of BO have been described, distinguished by their response to azithromycin: A responsive, neutrophilic, partially reversible form and a nonresponsive fibroproliferative variant.74,75 The association between BO and chronic GVHD has led to the hypothesis that host bronchiolar epithelial cells serve as target for donor cytotoxic T lymphocytes.76 Alternative explanations include recurrent aspiration of oral material due to esophagitis associated with GVHD, abnormal local immunoglobulin secretory function in the lungs, or unrecognized infection.69,76 The variations in histopathology, bronchoalveolar cell differential, clinical course, and the frequency of associated pulmonary infection suggest that the pathogenesis is multifactorial.76,77 BO in HSCT recipients resembles chronic allograft rejection in lung transplant recipients at immunological, pathological, and physiological levels.78 T-cell–mediated recognition of alloantigens expressed in the lung tissue plays a central role.68 In one study, none of the HSCT recipients with T-cell depletion developed BO.79
Clinical Findings
From several reports, the onset of BO occurs at a median 328 to 335 days and range 48 to 1690 days following HSCT.60,64,65,70,72,79 In the International Bone Marrow Registry of 6275 allogeneic HSCT recipients with leukemia, median time (range) from transplant to BO is 431 (65–2244) days.67 Twenty percent of the patients with BO had no respiratory symptoms at the time of the first abnormal pulmonary test. The absence of symptoms has been observed during the mild stage of BO.80 Typical respiratory symptoms include dry cough, dyspnea and exercise intolerance.70,72,81 The physical finding of wheezing may be detected in about 40%.70 Unlike COP, fever is absent in BO. Since these symptoms are nonspecific, a complete history, including prior medications and infections, and thorough physical examination focusing on signs of chronic GVHD should be obtained.
Diagnosis
The clinical criteria that have been most widely used for the diagnosis of BO in the HSCT population include FEV1/FVC <0.7 and FEV1 <75% of predicted; evidence of air trapping, small airway thickening, or bronchiectasis on HRCT of the chest (with inspiratory and expiratory cuts); residual volume >120% of predicted; and absence of infection in the respiratory tract.46,82 While definitive diagnosis ideally would include demonstration of histological evidence of BO, this almost always would require a surgical lung biopsy since the yield of transbronchial biopsy for this entity is extremely low. Because it is difficult to establish a histological diagnosis of BO by minimally invasive means, a surrogate functional diagnostic schema based on FEV1 decline was developed, referred to as BOS. BOS is defined as the presence of airflow limitation leading to a decline in FEV1 of at least 20% compared to baseline, in the absence of identifiable causes (e.g., infection). Based on the magnitude of decline in FEV1, BOS is classified into stages as follows: stage 1 (FEV1 66%–80% of peak posttransplant baseline), stage 2 (FEV1 51%–65% of baseline), and stage 3 (FEV1 <50% of baseline).83 This classification scheme was initially devised exclusively for lung transplant recipients and has not yet been widely applied to the HSCT population. Thus, its utility in defining severity or prognosis in this population remains to be established.
In HSCT recipients with suspected BO, laboratory evaluation should be undertaken to exclude infection and complications of GVHD. Complete blood count with differential, blood urea nitrogen, creatinine, total bilirubin, hepatic transaminases, immunoglobulin levels and subclasses, and urinalysis are recommended.76 Chest radiograph is usually normal or may show hyperinflation.70,78,84–86 Inspiratory and expiratory HRCT of the chest should be included as part of BO evaluation.80 HRCT of the chest typically shows mosaic attenuation due to air trapping (most pronounced on expiratory images), often accompanied by vascular attenuation, bronchiectasis, and bronchiolectasis (Fig. 95-2).78,85,87–89 GVHD is a risk factor for sinusitis in HSCT recipients.90 Since timely treatment of the sinusitis may lead to clinical improvement, radiological investigation including CT of the sinuses is recommended.76,90–93
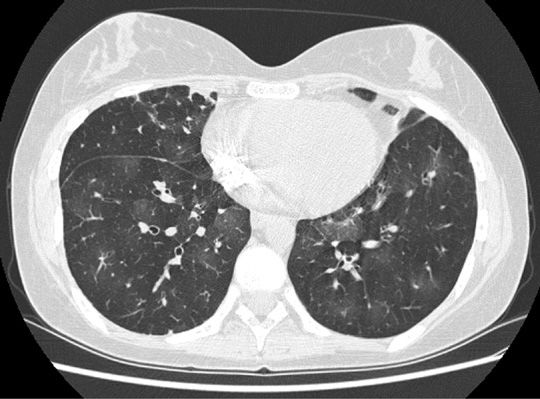
Figure 95-2 Computed tomography scan expiratory views of a patient with bronchiolitis obliterans following HSCT, showing mosaic attenuation associated with air trapping (darker areas), bronchial thickening, and mild bronchiectasis.
BAL is used in suspected BO to exclude infection and to assess the patterns of lung inflammation. In one study of seven HSCT patients with airflow obstruction, BAL showed predominantly neutrophilic inflammation in four, lymphocytic in two, and a mixed pattern in one.77 Because BO involves respiratory and membranous bronchioles, transbronchial lung biopsy is usually inadequate for diagnosis. Video-assisted thoracoscopic lung biopsy is a gold standard for definitive diagnosis, but is rarely pursued if other typical clinical–radiographic features are present.80 The pathology shows damage to the bronchiolar epithelium, fibrinous obliteration of bronchiolar lumen, inflammation between epithelium and smooth muscle, and pulmonary fibrosis (Fig. 95-3).78,81,94,95 Peribronchiolar inflammatory cellular infiltrates consisting of neutrophils and lymphocytes may be present.96
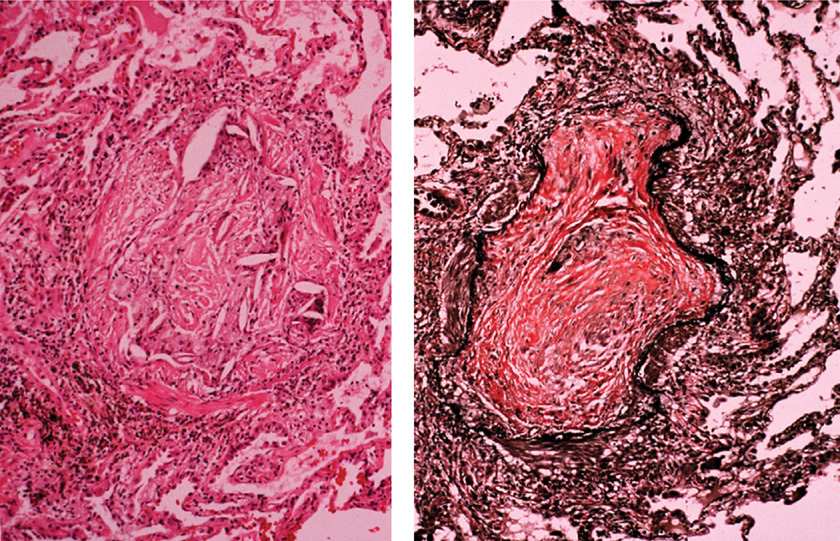
Figure 95-3 Lung pathology in bronchiolitis obliterans, showing inflammation and obliteration of the airway lumen, associated with fibrous connective tissue (hematoxylin and eosin stain, left; Verhoeff–Van Gieson elastic stain, right).
Treatment
There is insufficient evidence to support specific recommendations for the treatment of BO.68 First-line treatment usually involves corticosteroids.46 Prednisone 1 to 1.5 mg/kg/d, or its equivalent, is given for 4 to 6 weeks and if respiratory status remains stable, therapy is tapered and discontinued in 6 to 12 months. However, clinical responses are observed in only about 20% of patients and treatment success often does not persist.63,68,70,78,85,89,97 Immunosuppressive drugs used to treat BO associated with GVHD include cyclosporine, azathioprine, tacrolimus, and mycophenolate.46,76 The typical dose of azathioprine is 2 to 3 mg/kg/d, not to exceed 200 mg. Cyclosporine or tacrolimus dose is adjusted according to serum levels.58
Inhaled corticosteroids are used commonly to treat BO, but data are limited. A study comparing a combination of inhaled fluticasone, oral azithromycin, and montelukast in 9 patients with BOS against a historical control of 14 patients treated with high-dose corticosteroids showed no difference between the groups.98 Another study of 13 HSCT recipients with BO showed improvement in lung function and symptoms.99 These encouraging results provide potential for less toxic therapeutic options, but require confirmation in a larger group of patients with a longer follow-up period.
Macrolides have immunomodulatory effects that may halt disease progression in BO.100 Although the mechanism of action is not fully understood, it seems that it not only leads to a reduction of neutrophil inflammation,74,101 but also has prokinetic properties on the upper gastrointestinal tract, reducing gastroesophageal reflux, another risk factor for BO.102 Macrolides are commonly used to treat diffuse panbronchiolitis.103–107 There are case series documenting the beneficial role of macrolides for the treatment of BOS in lung transplant recipients.108 There are even less data addressing the role of macrolides in HSCT recipients with BO. In a series of eight patients treated with azithromycin 500 mg daily for 3 days, followed by 250 mg three times each week for 12 weeks, clinical improvement was achieved in seven patients.109 In a report of 10 HSCT recipients with BOS, azithromycin was not associated with improvement in lung function.110 Treatment of BOS in eight HSCT recipients with formoterol, azithromycin, and montelukast showed that this regimen may spare the use of high-dose systemic steroids.98 In a recent randomized double-blinded placebo-controlled trial of HSCT recipients with BOS, 10 were treated with oral azithromycin 250 mg daily while 12 were treated with placebo for 12 weeks.111 There were no significant differences in outcome between the two groups. Overall, there is insufficient evidence to substantiate the use of macrolides in the treatment of HSCT patients with BO.
Leukotrienes are eicosanoid lipid mediators that contribute to the inflammatory processes in the development of asthma, alveolitis, pulmonary fibrosis, and BO following lung transplantation, and promote bronchoconstriction, as well as eosinophil and neutrophil recruitment.112 There are limited data addressing the role of the leukotriene receptor antagonist montelukast in the treatment of BO. In a pilot study of 19 patients with refractory chronic GVHD, montelukast 10 mg daily was administered for a mean of 10 months.112 Overall improvement was observed in 15 of the 19 patients. In a prospective study comparing the combination of fluticasone, azithromycin, and montelukast against high-dose corticosteroids, there was no difference in lung function between the groups.98 However, with the montelukast combination, it was possible to taper systemic corticosteroids rapidly.
There are limited data regarding tumor necrosis factor (TNF) blockade in the treatment of BO. There is a case report of biopsy-proven BO after HSCT in a child who was treated with infliximab after failed corticosteroid therapy.113 The pulmonary symptoms and spirometric abnormalities resolved and the patient remained asymptomatic for several months.
The anti-CD4 monoclonal antibody, rituximab, has been used in refractory chronic GVHD. In one study, rituximab resulted in reduced systemic corticosteroid use.114 Improvement in lung function was noted in three of eight patients. In a phase II prospective multicenter study of HSCT recipients with steroid-refractory chronic GVHD, 37 patients were treated with weekly infusion of rituximab for 4 weeks followed by monthly infusion for 4 months.115 The treatment was complicated by infections and relapse. Only one of 11 patients showed partial response in the lung.
Extracorporeal photochemotherapy (ECP), or photopheresis, an immunomodulatory therapy developed for treating cutaneous T-cell lymphoma, has shown promise in treating chronic GVHD in uncontrolled studies.116 In this procedure, white blood cells are removed and exposed to ultraviolet radiation after pretreatment with a psoralen derivative, leading to cell death. In a retrospective study of 14 allogeneic HSCT recipients with extensive chronic GVHD, ECP was administered three times weekly on alternating days.116 Improvement in the pulmonary manifestations was noted in 1 of the 3 patients and systemic corticosteroid was tapered in 11 of the 14. In another study, nine allogeneic HSCT recipients with BO were treated with ECP.117 Six of nine (67%) patients responded after a median of 25 days. No ECP-related complications occurred. ECP stabilized rapidly declining pulmonary function tests in about two-thirds of patients with severe and refractory BO.
If otherwise successful HSCT is complicated by end-stage lung disease from BO, lung transplantation may be considered. There are insufficient data addressing this issue, with several single case reports.118–124 In a cross-sectional study of 313 lung transplant recipients, 3 were performed for BO in allogeneic HSCT recipients.123 Lung transplants from living donors have also been reported in this setting.121,124,125
Prognosis
The prognosis of BO in HSCT recipients is poor. Fewer than 20% improve and 65% die within 3 years of diagnosis.61,64,80,97,113 The rate of decline in FEV1 is widely variable and rapid deterioration is associated with increased mortality.70 Despite treatment, improvement in lung function is noted in only 8% to 20%.58 The reported case fatality rates vary widely, ranging from 14% to 100% with a mean of 61%.58 In one study of allogeneic HSCT recipients with GVHD, the 3-year mortality rate of those with BO was 65% compared to 44% of those without BO.70 In a more recent study, the 5-year survival of HSCT recipients with BO from the time of diagnosis was 45.4%, significantly less than those without (77.5%, p < 0.001).65 The mortality of BO has not improved over the last two decades.80
 PERI-ENGRAFTMENT RESPIRATORY DISTRESS SYNDROME
PERI-ENGRAFTMENT RESPIRATORY DISTRESS SYNDROME
PERDS is the pulmonary manifestation of engraftment syndrome in HSCT recipients.126 The engraftment syndrome is characterized by skin rash, noninfectious pulmonary infiltrates, fever, diarrhea, and capillary leak during the peri-engraftment period.127–129 Engraftment syndrome develops in 7% to 53% of HSCT recipients.127,128,130,131 Among 152 autologous HSCT recipients supported by either granulocyte or granulocyte macrophage colony–stimulating factors, engraftment syndrome developed in 20 (13%).132 Although most of the studies reporting engraftment syndrome are in autologous HSCT recipients, it also occurs after allogeneic transplantation.133,134 In two recent studies, pulmonary manifestations were reported in 18 of 61 (30%) patients with engraftment syndrome.127,131
Frequency, Risk Factors, and Pathogenesis
There is only one study that addressed PERDS specifically and reported an occurrence in 4.6% of 416 autologous HSCT recipients.126 Using similar criteria in the same institution, a recent study reported PERDS in 52 of 1243 (4.2%) autologous HSCT recipients.12
Earlier studies of the engraftment syndrome noted possible relationships with the underlying disease, therapies prior to transplant, stem cell dose and source, growth factor use, amphotericin administration, and rate of engraftment.130,133,135 In one study focusing specifically on PERDS, there was no association with demographics, type and stage of underlying disease, conditioning regimen, graft source, use of cytokines either for stem cell mobilization or after transplant, viral serologies, pretransplant pulmonary function, or laboratory values.126
The pathogenesis of PERDS is not well defined, but is believed to represent a complex interaction between conditioning-related endothelial damage and the cytokine release associated with neutrophil and lymphocyte recovery.126 Tissue infiltration by neutrophils has been shown to occur earlier than their appearance in the peripheral blood.136
Clinical Findings
In 19 patients with PERDS, median duration from transplant to onset of symptoms or radiographic pulmonary infiltrates was 11 days (range, 4–25).126 The median duration to neutrophil engraftment was also 11 days after transplant (range, 8–25). Symptoms and signs occurred within 5 days of neutrophil engraftment. Dyspnea was the initial symptom in all patients, and fever was present in 12 patients (63%).
Diagnostic Evaluation
PERDS is considered in the presence of fever (>38.3°C) and pulmonary injury with evidence of hypoxemia (SaO2 <90%) and/or pulmonary infiltrates on chest radiograph, in the absence of clinical cardiac dysfunction or infection, all occurring within 5 days of neutrophil engraftment (defined as absolute neutrophil count of more than 500/mL on consecutive days).126 The median white blood cell count at the time of symptom onset is 1.3 × 109/L (range, 0.2–29.6) and the median neutrophil count is 0.7 × 109/L (range, 0.1–28.6). Chest radiographs show bilateral infiltrates in the majority. In one study, BAL was performed to exclude pulmonary infections, but in a more recent study, BAL was not performed if noninvasive evaluation was considered adequate.12 Transbronchial lung biopsy cannot be performed safely in most HSCT recipients during the peri-engraftment period because of thrombocytopenia. Surgical lung biopsy may show diffuse alveolar damage, but is rarely necessary.126
Treatment
In the only study focusing on PERDS, 11 patients received high-dose corticosteroid therapy, including 5 of the 6 who required mechanical ventilation.126 Ten patients experienced clinical improvement, which occurred within 24 hours in five cases, and over 2 to 4 days in the remainder. The rapid response to corticosteroid treatment and the fact that such a therapy was delayed until after intubation in all the mechanically ventilated patients suggested a therapeutic benefit. Among the eight patients who did not receive steroids, one required mechanical ventilation at the onset of symptoms, and subsequently died.126 The other seven patients recovered and were discharged from the hospital. There was no correlation between gender, age, source of stem cells, mononuclear cell count of the graft, use of posttransplant growth factors, WBC at the onset of symptoms, rate of rise in WBC, amphotericin use, or the need for mechanical ventilation and posttransplant mortality.126 The mean absolute neutrophil count was significantly higher among patients with evidence of alveolar hemorrhage by BAL. Unlike DAH and IPS, only about one-third of HSCT recipients with PERDS require ICU admission and mechanical ventilation.
 ACUTE PULMONARY EDEMA
ACUTE PULMONARY EDEMA
Acute pulmonary edema is common during the neutropenic phase and likely represents a combination of cardiogenic and noncardiogenic factors. Noncardiogenic pulmonary edema has many contributory factors including total-body radiation, induction drugs, and septic episodes. These factors likely damage the capillary endothelium, leading to radiographic findings similar to hydrostatic edema. Patients with hydrostatic pulmonary edema have often received high volumes of fluid for medications, total parenteral nutrition, and multiple blood product transfusions. The heart may also be compromised by chemotherapeutic agents such as Adriamycin and high-dose cyclophosphamide used during induction. In a recent study of 1243 autologous HSCT recipients, acute pulmonary edema was reported in 4.7%.12 Patients develop weight gain and dyspnea, and bibasilar crackles. Radiographic findings of vascular and hilar indistinctness, symmetric ground-glass/consolidative opacities, enlarged heart, small pleural effusions, and subfissural thickening are typically seen. While pulmonary edema is usually diagnosed by plain chest radiograph, CT findings include prominent pulmonary vessels, interlobular septal thickening, ground-glass attenuation, and pleural effusions. Acute pulmonary edema usually can be prevented and treated with fluid restriction and diuretics.
 IDIOPATHIC PNEUMONIA SYNDROME
IDIOPATHIC PNEUMONIA SYNDROME
Important aspects of the idiopathic pneumonia syndrome are considered below, including frequency, risk factors, clinical findings, treatment, and prognosis.
Frequency
The term IPS has been used for many years to describe lung injury without infectious etiology in HSCT recipients. In a 1985 review of pulmonary complications in 4500 HSCT recipients, Krowka et al. reported a 35% frequency of idiopathic pneumonia.12,137 Another study from a major HSCT center found a rate of 7.3%.138 The wide variation in the reported incidence of IPS was partly due to the lack of uniform definition and diagnostic criteria. Diagnostic criteria were addressed in 1993 by a workshop sponsored by the National Heart, Lung, and Blood Institute.139 This workshop defined IPS as widespread alveolar injury in the absence of lower respiratory tract infection.139 The definition has been updated recently.140 The current criteria for IPS include evidence of widespread of alveolar injury, absence of active lower respiratory tract infection, and exclusion of cardiac dysfunction, renal failure, or fluid overload.140 Based on data compiled from several studies of 4496 HSCT recipients, 449 cases of IPS (10%; range 2%–17%) were reported.138,140–149
Risk Factors
The probability of developing IPS increases with the number of risk factors.148 Patients transplanted for aplastic anemia have low risk for IPS.150 With the advent of prophylaxis for CMV infection, IPS has become a relatively more frequent cause of interstitial pneumonia.151 Using multiple logistic regression analysis, Kantrow et al.138 did not find significant difference in the rate of IPS between autologous and allogeneic HSCT recipients. However, a review of selected studies showed 36 of 617 autologous HSCT recipients (5.8%) developed IPS compared to 380 of 3569 allogeneic HSCT recipients (10.6%), a significant difference.58
Pathogenesis
Although the pathogenesis of IPS is not well defined, lung injury, inflammation, and cytokine release are implicated.58 Pulmonary vascular endothelial cell damage, mediated by TNF-α, is linked to the development of experimental IPS.140,152 Another potential mechanism is parenchymal damage from previous chemoradiation therapy, GVHD, undiagnosed infection, and excessive recruitment and activation of inflammatory cells.138 The occurrence of IPS after autologous BMT suggests that the pretransplant conditioning regimens, rather than GVHD and CMV infection, are likely to be culprits.153 Latent and unrecognized infections such as human herpes virus-6 (HHV-6) and systemic activation of inflammatory cytokines during sepsis may also be responsible for lung injury.138,154–157 Cell-mediated immune injury during GVHD reactions is another possible mechanism.138 Further, IPS may be the result of persistent proinflammatory events and oxidant responses.158,159
Clinical Findings
The median time of onset of IPS is 19 days, range 4 to 106 days, after transplant.140 Despite the variable onset, the majority of patients present within the first 120 days following HSCT.138,144 The clinical presentation of IPS includes dyspnea, dry cough, hypoxemia, and nonlobar radiographic infiltrates.139 The spectrum is broad, ranging from acute respiratory failure to incidental radiographic abnormalities. Because IPS mimics infectious pneumonia, the majority of patients are on antibiotics at the time of diagnosis.
Diagnostic Evaluation
Clinical presentation and radiographic findings cannot be used to differentiate between patients with infectious and idiopathic pneumonia. Pulmonary function testing and computed tomography (CT) of the chest are also nonspecific.138 More than 90% of patients with IPS have diffuse infiltrates on chest radiograph.160 Based on the IPS criteria, PERDS and DAH are considered to be subsets of IPS. There is overlap between PERDS and DAH; however, the clinical course and response to corticosteroid therapy differ between these conditions.
Infection should be excluded before the diagnosis of IPS is made. In earlier studies, IPS was diagnosed histologically when biopsy or autopsy of lung tissue showed inflammation without any histological or microbiological evidence of infection.144,149,150 In a study from Fred Hutchinson Cancer Research Center, 80% of IPS cases were diagnosed without tissue and with infection excluded by BAL, 4% required lung biopsy, and 16% were diagnosed at autopsy.138 Although the NHLBI workshop accepted BAL as the main method for the exclusion of infection and the diagnosis of IPS, the limitations of this approach in excluding invasive fungus, neoplasms, and other abnormalities of potential therapeutic or prognostic importance are well recognized.138 When pursued, lung biopsies of patients with IPS show diffuse alveolar damage, organizing or acute pneumonia, and interstitial lymphocytic inflammation.138,143
Treatment
There are scarce data addressing the treatment of IPS in HSCT recipients. One study reported three patients with IPS who responded to treatment, which included corticosteroids.138,143 Despite treatment with methylprednisolone at 1 to 2 mg/kg/d, studies with larger sample sizes have not shown any outcome benefit.138,160 There are reports of IPS responding to etanercept.161–163 Currently, the only accepted treatments are supportive care, and prevention and treatment of infection. Lung transplant offers a potential therapeutic option for patients who develop respiratory failure despite treatment, but this option is limited to the rare circumstances of a patient without other comorbidities and for whom the transplant was considered curative to have definitively eradicated the underlying disease process.120
Prognosis
The clinical course of IPS is commonly complicated by infections, including viral and fungal pneumonias.138,160 Other complications include pneumothorax, pneumomediastinum, subcutaneous emphysema, and pulmonary fibrosis. Autoimmune polyserositis involving the pleura and pericardium has also been reported.164
Patients often develop severe hypoxemia and require assisted ventilation.165 In a review of selected studies, the overall mortality of 388 HSCT recipients with IPS was 74%, with a range of 60% to 86%.138,144,148–150,160 The 1-year survival rate is less than 15%.138,160 Infectious complications and nonpulmonary organ failure contribute to the high mortality rate.138,160 For those who require mechanical ventilation, the hospital mortality may exceed 95%.138
 DIFFUSE ALVEOLAR HEMORRHAGE
DIFFUSE ALVEOLAR HEMORRHAGE
Although pulmonary infections can cause alveolar hemorrhage, the term DAH in the HSCT recipient is reserved for alveolar hemorrhage of noninfectious etiology.
Frequency
The frequency of DAH has varied among reported series because of differences in patient mix and diagnostic criteria.166 Factors that influence the incidence of DAH have changed over time and among HSCT centers. In a review of several studies that included 3806 HSCT recipients, the cumulative frequency of DAH was 5%, ranging from 2% to 14% in the individual series.166 Among 692 HSCT recipients who underwent bronchoscopy, DAH was reported in 97 (14%), with a range between 1% and 23%.126,167–173 In patients admitted to the intensive care unit for respiratory failure, the prevalence of DAH may exceed 40%.174,175 DAH has been reported in approximately 10% of autopsies of HSCT recipients.16,17
Risk Factors
Pretransplant chemotherapy and conditioning regimen, total-body irradiation, thoracic irradiation, and older age are associated with DAH.9,153,176–179 There are conflicting data regarding solid tumors as a risk factor for DAH.166,180,181 Although DAH occurs in both autologous and allogeneic HSCT recipients, the initial studies included mostly autologous recipients.153 In data from several reports, however, no significant difference was found in the incidence of DAH between autologous and allogeneic HSCT recipients.166 Pulmonary function tests have shown no association between the development of DAH and pretransplant PFTs.153 Pretransplant bronchoscopy has shown a higher number of bronchial neutrophils and eosinophils in patients who develop DAH after HSCT compared with those who do not.182 White blood cell recovery and renal insufficiency, but not prolonged prothrombin, partial thromboplastin time, or low platelets, are associated with the development of DAH.9,153 Although most patients with DAH have thrombocytopenia, DAH is not corrected by platelet transfusion.153
Pathogenesis
Various conditions, including infections, mitral valve disease, systemic vasculitides, collagen vascular diseases, drugs, and anticoagulation have been implicated as causing alveolar hemorrhage in non-HSCT recipients. The etiology and pathogenesis of DAH in the HSCT recipient have not been clearly established. Lung tissue injury, inflammation, and cytokine release are implicated.166
Pretransplant high-dose chemotherapy, thoracic or total-body radiation, and undocumented infections may be responsible for the initial injury to lung tissue. Vascular endothelial swelling and thrombi are found in the autopsies of HSCT recipients with acute hemorrhagic pulmonary edema.183 The incidence of pulmonary hemorrhage is high in HSCT recipients with GVHD.179 In addition to the toxicity from therapy for GVHD, antigen-specific injury to endothelium may be a contributing factor to the development of DAH.184
Vasculopathy of small muscular arteries and thrombotic microangiopathy have been reported in HSCT recipients with DAH.185 The vasculopathy manifests as concentric intimal or medial hyperplasia with luminal narrowing, prominent myxoid change, extravasated red blood cells, and the presence of foamy histiocytes. Thrombotic microangiopathy has also been associated with DAH, characterized by fragmented erythrocytes on peripheral smears, decreased hemoglobin and platelet counts, refractoriness to platelet transfusions, and the absence of disseminated intravascular coagulation.185
Inflammatory cells are likely to play a role in the development of DAH, and models are characterized by increase in the alveolar leukocytes, platelet microthrombi, damage of alveolar endothelial and epithelial cells, increased turnover rate of alveolar cells, and an increase in the cell number and protein content of the BAL.186 Pretransplant bronchoscopy has shown increased bronchial inflammatory cells in patients who develop DAH after HSCT, suggesting that bronchial inflammation precedes alveolar inflammation.182,187 The initial injury is compounded by damage related to the return of inflammatory cells to the lung coincident with marrow recovery.188 Even in the presence of peripheral leukopenia, neutrophils are seen in the lower respiratory tract of HSCT recipients at the time of DAH.166 One-third of autologous HSCT recipients with PERDS have DAH.126 Among the patients with PERDS, those with DAH have higher absolute neutrophil counts than those without DAH.
In allogeneic transplants, donor T cells react to host alloantigens, proliferate, and secrete inflammatory mediators. This response may be amplified by the release of endotoxin from the gut after injury from mucositis or GVHD. It is suggested that the pathophysiology of acute GVHD is a cytokine storm of inflammatory mediators.189 In autologous HSCT recipients, the generation of cytokines is self-limited and resolves in 7 to 10 days.189
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


