Histiocytic disorder
Morphology
Immunophenotype
Reactive histiocytic conditions
Non-specific interstitial pneumonitis
Polymorphous infiltrate containing macrophages
Bronchiolar obstruction
Foamy cells
Healing process
Infections
Mycobacteria infections
Foamy histiocytes
PAS+/Ziehl+
Whipple disease
Foamy histiocytes
Antibody to T whipplei
PAS+/Gram+
Malakoplakia
Michaelis-Gutmann bodies
PAS+/Perls+/Von Kossa+
Crystal-storing histiocytosis
Immunoglobulin crystals within macrophages
CD68+
Light-chain restriction in macrophages and plasma cells
Storage diseases
Gaucher’s disease
PAS+, wrinkled paper aspect
CD68+
Niemann-Pick’s disease
Foamy cells
CD68+
Primary histiocytic disorders
Langerhans cell histiocytosis (LCH)
Folded nucleus, pale cytoplasm
CD1a+/CD207+/S100+/CD68±BRAF(V600E) ±
Eosinophils
Erdheim-Chester disease (ECD)
Round or oval nucleus
CD1a−/CD207−/S100±/CD68+/FXIIIa+BRAF(V600E) ±
Pale or foamy cytoplasm
Multinucleate giant Touton cells
Lymphocytes, plasma cells
Rosai-Dorfman disease (RDD)
Round or oval nucleus
CD1a−/CD207−/S100+/CD68+/FXIIIa−
Pale or foamy cytoplasm
Multinucleate giant cells
Emperipolesis
Lymphocytes, many plasma cells
Histiocytic malignant neoplasms
Histiocytic sarcoma
Pleomorphism
CD68+/CD163+CD1a−/CD21−/CD35−Myeloperoxidase−
Atypical nucleus
Mitoses
Follicular dendritic cell sarcoma
Spindle shaped tumor cells
CD23+/CD21+/CD35+
Ovoid or elongated nuclei
Storiform pattern
Interdigitating dendritic cell sarcoma
Spindle shaped tumor cells
S100+/CD68−/CD163−CD1a−/CD21−/CD35−
Ovoid or folded nuclei
Primary Langerhans cell sarcoma
Pleomorphism
CD1a+/CD207+/S100+
Atypical nucleus
Mitoses
Erdheim-Chester Disease (ECD)
A Increasingly Well Recognized Multisystemic Disease: A General Review (Apart from the Lung)
Historical Considerations
Erdheim-Chester disease (ECD) is a rare, non-Langerhans form of histiocytosis of unknown origin. It was first described as “lipoid granulomatose” by Jakob Erdheim’s student William Chester, in 1930 [1]. More than 500 distinct cases have since been reported [2]. ECD is characterized by the xanthomatous or xanthogranulomatous infiltration of tissues by foamy histiocytes, “lipid-laden” macrophages or histiocytes, surrounded by fibrosis [3, 4]. It can be distinguished from Langerhans cell histiocytosis (LCH) on the basis of the immunohistologic characteristics of histiocytes, as the cells stain positive for CD68 and negative for CD1a in ECD. In most cases (80 %), staining for the S-100 protein is also negative.
Two signs highly evocative of ECD are the nearly constant tracer uptake by the long bones on 99Technetium bone scintigraphy and a “hairy kidney” appearance on abdominal CT scan present in approximatively 50 % of cases. Diagnostic criteria classically used are mentioned in the “box criteria”.
Demographic Characteristics
Based on our experience with 75 patients, published in 2012, there is a strong male predominance in this disease, with 73 % of patients being male and only 27 % female [5]. Mean age at diagnosis in the two large series published was relatively stable: 55 years ±14 (range, 16–80 years) in the 2011 series [2]. ECD is far less frequent in children, as only eight pediatric cases have been reported, all with no cardiac involvement [6, 7].
Clinical Phenotypes
ECD is a truly systemic disease, with diverse signs, including skeletal involvement with bone pain, exophthalmos, diabetes insipidus, xanthelasma, interstitial lung disease, bilateral adrenal enlargement, retroperitoneal fibrosis with perirenal and/or ureteral obstruction, renal impairment, testis infiltration, central nervous system (CNS) and/or cardiovascular involvement [2, 4]. The frequency of the main clinical and radiological characteristics in ECD are presented in Table 14.2. The baseline clinical evaluation recommendations for patients diagnosed with ECD are also listed in a box.
Table 14.2
Frequency of the main clinical and radiological characteristics in Erdheim-Chester disease
From the literature, % | Personal experience, %a | |
|---|---|---|
Bone pain | 50 | 40b |
Peri-aortic infiltration | 60 | 66 |
“coated aorta” (sheathing of the whole thoraco-abdominal aorta) | 30 | 23b |
Pericardial involvement | 45 | 42 |
Exophthalmos | 27 | 25 |
Diabetes insipidus | 27 | 25b |
Xanthelasmas | 19 | 28 |
“hairy kidney” aspect | ND | 68 |
CNS involvement | 15–25 | 51 |
Pulmonary involvement | 22 | 43 |
Death | 60 | 26 |
The clinical course of ECD depends largely on the severity and distribution of the disease, and may range from asymptomatic bone lesions to multisystemic, life-threatening forms with a poor prognosis.
Clinical Vignette
The patient was a 55 year-old man who was diagnosed with pituitary adenoma in 2012 after a 5 year-history of diabetes insipidus. At that time, he was admitted in Intensive Care Unit for respiratory failure, which was attributed to an infectious disease. In March 2013, he presented with a second episode of respiratory failure and chest physicians were puzzled by an unusual septa infiltration on chest CT scan (see Clinical Vignette figure). Systemic explorations revealed bilateral symmetric femoral and tibial bones, lung and suprasellar involvement, with no cardiac or retroperitoneal infiltration. Diagnosis of ECD was finally made from the xanthelasma biopsy which demonstrated foamy CD68+ CD1a− histiocytes, in which BRAFV600E mutation was found.
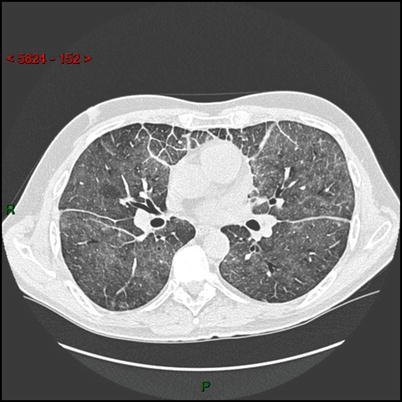
HRCT scan of the lung: axial image showing diffuse ground glass opacities, thickening of interlobular septa in the anterior parts of the lungs and thickening of the fissures due to a thickening of the subpleural interstitium.
Treatments
Until 2005, treatments for ECD included steroids, cytotoxic agents [8] and double autologous hematopoietic stem-cell transplantation [9, 10]. The efficacy of these treatments was difficult to assess, because each individual treatment had been used in only a few patients or in combination with other drugs and follow-up periods were often short. Braiteh et al. treated three ECD patients with interferon alpha (IFNα) and reported a rapid, substantial and durable regression of retro-orbital infiltration and gradual improvements of bone lesions, pain and diabetes insipidus [11]. However, we found, in eight patients with ECD, that the efficacy of low-dose IFNα (3 MU × 3/week) differed between patients and with the disease sites involved [12]. Moreover, symptoms did not always respond to IFNα at such doses, particularly in patients with severe multisystemic forms of ECD (CNS and cardiovascular involvement) [2]. We therefore recommend the use, if possible, of higher doses, of up to 9 MU × 3/week, which may be more effective for the treatment of meningeal infiltrations, sub- and retrosellar masses, pericardial and pseudoatrial infiltrations. Long-term treatment should be considered, although it may be difficult to achieve due to side effects, such as depression and fatigue. IFNα has given much less convincing results for the treatment of pseudodegenerative forms of ECD with cerebellar involvement (similar to those observed in LCH).
IFNα appears to be a valuable first-line therapy for the prolonged treatment of ECD. A recent survival analysis revealed that treatment with IFNα and/or PEGylated IFNα was a major independent predictor of survival in a series of 53 patients (HR = 0.32; 95 % CI, 0.14–0.70; P = 0.006) [2]. The current therapeutic approach involves the initiation of treatment with PEGylated forms of IFNα, which are often better tolerated in the long term.
In 2010, there were several reports suggesting that imatinib mesylate was an effective treatment for histiocytoses [13], although a preliminary experience, with six ECD patients, was disappointing [14]. In the same year, recombinant human interleukin-1 receptor (anakinra) was also reported as a promising treatment in two patients with ECD, but these patients had no cardiovascular or CNS involvement [15]. Given the efficacy of cladribin for treating LCH, this drug is also a potentially interesting treatment for sites of ECD in the CNS that are refractory to IFNα [8]. Dagna et al. recently reported the key role of TNFα in ECD and the efficacy of infliximab in two patients with this disease [16].
Pathophysiology
The understanding of the pathogenesis of ECD has improved since 2006. In an immunohistochemical study of three patients, Stoppacciaro et al. showed that a complex network of cytokines and chemokines regulates histiocyte recruitment and accumulation in the lesions [17]. Dagna et al. recently assessed both spontaneous and stimulated cytokine production by mononuclear cells from the biopsy fragments of a single patient [18]. This study revealed that tumor necrosis factor α (TNFα) was produced after stimulation and described the spontaneous secretion of IL-6 and IL-8, a known chemoattractant for polymorphonuclear cells and monocytes. Aouba et al. showed, in two patients, that targeting the IL-1 pathway might be an appropriate strategy [15]. We recently analyzed 23 cytokines production in serum samples obtained from a large cohort of ECD patients [19]. Our data revealed intense systemic immune activation in these 37 patients, mostly involving IFNα, IL-1/IL1-RA, IL-6, IL-12 and MCP-1. These findings further highlight the importance of the disruption of the systemic immune Th-1–oriented response associated with this condition, providing clues to more focused therapeutic agents.
Impact of BRAF Mutations on Management
The mitogen-activated protein kinase (MAPK) has been implicated in the pathophysiology of many cancers, especially melanoma, lung and colorectal cancers. Under physiologic conditions, the RAS-RAF-mitogen activated protein kinase (MEK)-mitogen-activated protein kinase (ERK) signaling cascade interaction is initiated by ligation of a receptor-linked tyrosine kinase by its corresponding growth factor. BRAFV600E mutation is responsible for a constitutively active kinase. Vemurafenib – a selective low molecular weight BRAF kinase inhibitor effective for the treatment of metastatic melanoma, including cerebral metastases – has been shown to be highly effective for treating bone, retro-orbital, retroperitoneal and cardiac lesions in three ECD patients with mutations.
In 2012, we showed that BRAF V600E gain-of-function mutations were present in 57 % of LCH cases and 54 % of ECD cases, but not in other types of histiocytosis, confirming the clonal nature of LCH and ECD in large subsets of patients [20]. Targeted therapy with an inhibitor of mutated BRAF — vemurafenib, a newly approved selective low molecular weight BRAF kinase inhibitor — improves survival in patients with melanoma including cerebral metastases. Vemurafenib selectively suppresses proliferation in tumour cells expressing mutated BRAFV600E proteins. We used vemurafenib to treat three patients with multisystemic and refractory ECD carrying the BRAF V600E mutation, two of whom also had skin or lymph node LCH involvement [21]. Vemurafenib treatment led to rapid, substantial, clinical and biological improvement in all patients, and the tumor response was confirmed by positron emission tomography (PET), computed tomography (CT) and/or magnetic resonance imaging (MRI) 1 month after treatment initiation. The treatment was still effective after 4 months of follow-up, although persistent disease activity was still observed. Such short study was proof-of-concept in showing that BRAF mutation may play a direct role in the physiopathology of the disease. We believe that treatment with vemurafenib should be considered for patients with severe, refractory BRAF V600E histiocytosis, particularly if the disease is life-threatening. An appropriate dosing schedule and treatment duration remain to be determined, but these findings provide compelling evidence that BRAF inhibition is rapidly effective against BRAF V600E -associated ECD. The long-term efficacy of BRAF inhibition in ECD should also be studied, as secondary resistance develops in almost all cases of BRAF V600E -associated melanoma.
Pulmonary Involvement in ECD
Prior to the 2009 review of the pulmonary aspects of ECD, only a few studies had assessed the frequency of pulmonary involvement in ECD. Evidence of such involvement was found in eight (14 %) of the 59 patients reported by Veyssier-Belot et al. [3]. In a previous review, 41 (23 %) of the 176 cases reported in articles published in English (up to June 2003) were also found to display pulmonary involvement [22].
In a subsequent MEDLINE search, we identified 72 (23 %) cases of clear-cut pulmonary involvement in 319 patients published between 1930 and November 2008 (23, Table 14.3). The prognosis was grim in some of these cases of pulmonary involvement sometimes complicated with fatal respiratory failure due to pulmonary fibrosis. However, data regarding pulmonary involvement were incomplete for many cases in these reports, highlighting the lack of recognition of pulmonary signs in ECD patients.
Table 14.3
Main characteristics of the 72 patients with pulmonary involvement of ECD published in the medical literature
Case number | Authors | Year | Sex/age | Original case nba | Respiratory clinical manifestations | Respiratory radiological findings | Histology | Follow-up | Others organs involved |
|---|---|---|---|---|---|---|---|---|---|
1 | Chester | 1930 | F/44 | 1 | Dyspnea | NA | Autopsy | Death by heart failure | Eye, liver, skin |
2 | Heine | 1934 | M/50 | NA | NA | Autopsy | NA | CNS, eye | |
3 | Masshoff | 1948 | F/45 | NA | NA | Autopsy | Death by dehydration | CNS | |
4 | Cavanagh | 1954 | F/64 | 1 | Dyspnea | Opacities of upper lobes | Autopsy | Death by heart failure | Bone, CNS, kidney, large-vessels, skin |
5 | F/49 | 2 | None | NA | Autopsy | Death | Bone, CNS, kidney, large-vessels, skin | ||
6 | Elian | 1969 | M/40 | NA | NA | Autopsy | Death | Skin, CNS | |
7 | Jaffe | 1972 | F/54 | NA | NA | NA | NA | NA | |
8 | Resnick | 1982 | M/76 | 1 | NA | Diffuse fibrosis | Autopsy | Death by sepsis | Bone, kidney |
9 | Alper | 1983 | M/65 | Dyspnea | Diffuse fibrosis, pleural effusion | Autopsy – TBB – lung | Death by heart failure | Eye, liver, spleen, skin | |
10 | F/40 | NA | ILD, pleural effusion | Autopsy | Death by cerebral hemorrhage | CNS, eye, bone, skin | |||
11 | Palmer | 1984 | M/51 | None | ILD | Retroperitoneal biopsy | Alive at 2 years | Bone, kidney, eye, liver | |
12 | Sherman | 1985 | M/70 | 1 | Dyspnea | NA | TBB – lung | Death | Eye |
13 | Freyschmidt | 1986 | F/60 | NA | ILD | Autopsy | Death within 3 weeks | NA | |
14 | Miller | 1986 | M/44 | Cough, pleural pain | Pleural thickening | Skin, bone, liver | NA | Bone, skin, liver, spleen | |
15 | Sandrock | 1989 | M/21 | 1 | Dyspnea, pericarditis | Pleural infiltration | Pericardium | Alive | Heart, skin, bone, liver, spleen |
16 | Globerman | 1991 | M/7 | None | Perivascular infiltration | NA | Alive | NA | |
17 | Fink | 1991 | M/76 | NA | Pleural effusion | Autopsy | Death by myocardial infarction | CNS, eye, bone | |
18 | Kujat | 1991 | M/53 | NA | ILD | Retroperitoneal biopsy | NA | CNS, kidney, skin | |
19 | Schields | 1991 | M/38 | 1 | NA | Pleural thickening | Eye | Death by heart and renal failures | Eye, kidney, skin |
20 | Athanasou | 1993 | M/72 | NA | NA | Lung | NA | CNS, heart, bone | |
21 | Remy-Jardin | 1993 | F/50 | Dyspnea, crackles | ILD | TBB, lung, bone | Respiratory involvement worsening. | Eye, skin, bone, CNS, kidney, liver, spleen, large-vessels | |
Madroszyk | 1994 | ||||||||
Caparros-Lefebvre | 1995 | 3 | Death by CNS involvement | ||||||
Farre | 1995 | 2 | |||||||
22 | Veyssier-Belot et al. | 1996 | F/50 | 3 | Dyspnea | ILD | TBB, bone | Death by CNS involvement | Bone, eye, CNS |
23 | Devouassoux | 1998 | F/41 | Dyspnea | ILD | Lung | NA | Bone | |
24 | Egan et al. | 1999 | F/62 | 1 | Cough, dyspnea | NA | Lung CD 68+/CD1a− | Death by pulmonary fibrosis | CNS |
25 | F/46 | 2 | Cough, dyspnea | NA | TBB−/bone+/skin+ | Stability | Skin, adrenal glands | ||
26 | F/25 | 3 | Cough, dyspnea, pleural pain | NA | Lung CD68+/CD1a−, skin | NA | CNS, skin | ||
27 | F/65 | 4 | Acute respiratory failure | NA | Lung CD 68+/CD1a− | NA | NA | ||
28 | H/70 | 5 | Dyspnea | NA | Lung CD 68+/CD1a− | Stability | CNS | ||
29 | Rush | 2000 | M/71 | 1 | Dyspnea | ILD, pleural thickening | Lung, bone | Death | Bone |
30 | M/63 | 2 | Dyspnea, cough | Cysts, ILD, pleural thickening | Lung, osteomedullar biopsy | Death by respiratory failure | Bone, liver, spleen | ||
31 | F/69 | 3 | Dyspnea | Pleural effusion and thickening, ILD | Lung, retro-orbital, autopsy | Death by respiratory failure | Bone, eye | ||
32 | Wittenberg
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|