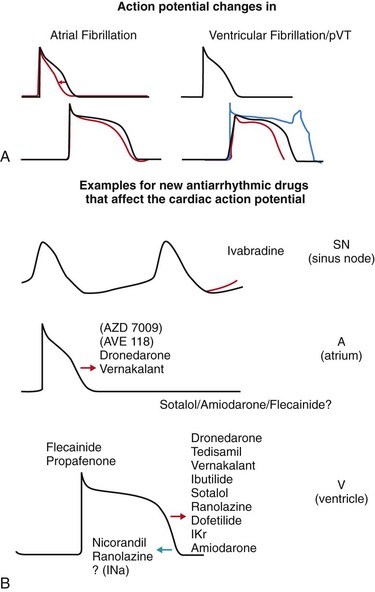
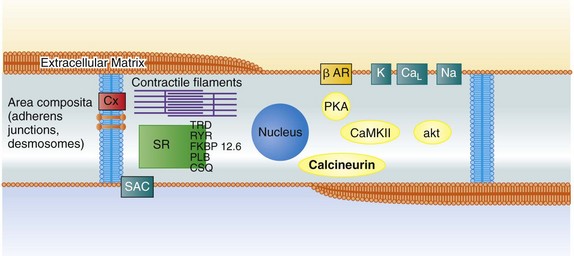
112 Although ion channel–blocking antiarrhythmic drugs such as quinine and digitalis glycosides have been among the first compounds that were evaluated in controlled clinical trials,1 most antiarrhythmic drugs in clinical use today have been developed for different indications than those for which they are used: Many drugs that are currently primarily used in atrial fibrillation were initially developed to prevent sudden death. Furthermore, antiarrhythmic drugs are currently often part of a “hybrid” therapy: used—for example, in patients with defibrillators are treated to reduce shocks, or patients with atrial fibrillation receive antiarrhythmic drugs to reduce recurrences of atrial fibrillation within a therapeutic concept that also makes use of catheter ablation. As one of several consequences, complete prevention of arrhythmic events is much less needed than it was when the current drugs were initially developed. Many medications can have antiarrhythmic effects, and the complex mechanisms that initiate atrial fibrillation and sudden death are discussed elsewhere in this book. Understanding these mechanisms can provide new targets for antiarrhythmic drugs (Figure 112-1). Classical, ion channel–blocking antiarrhythmic drugs have a few main electrophysiological mechanisms of action. Figure 112-1 Targets for antiarrhythmic drug treatment for the prevention of atrial fibrillation and sudden cardiac death. Shown is a histologic scheme of a cardiomyocyte with some key proteins that might represent targets for antiarrhythmic drug therapy. Antiarrhythmic drug classes are indicated within the scheme to illustrate potential sites of action. CaL, L-type calcium channel; Na, sodium channel; K, potassium channels; Cx43, connexin43; CaMKII/akt/calcineurin, examples for relevant protein kinases; SAC, stretch-activated channel; SR, sarcoplasmic reticulum; SERCA2A, sarco-endoplasmic reticulum calcium-dependent ATPase type 2A; FKPB 12.6, FKPB 12.6, an accessory protein to the ryanodine receptor; RYR, ryanodine receptor; PLB, phospholamban; TRD, triadin; CSQ, calsequestrin. Most antiarrhythmic drugs prolong the cardiac action potential by inhibiting repolarizing potassium currents. This effect is reflected by the QT-prolonging effect in the electrocardiogram (ECG) for ventricular repolarization and has been demonstrated by invasive electrophysiological measurement of action potential duration and refractoriness in the atria (Figure 112-2). Action potential prolongation is believed to be the main antiarrhythmic mechanism of current antiarrhythmic drugs, and reverses the main electrophysiological effect associated with electrical remodeling in atrial fibrillation.2 Unfortunately, prolongation of the ventricular action potential also appears to be one of the main mechanisms of drug-induced ventricular proarrhythmia by provoking torsades de pointes tachycardia.3 Whereas many new antiarrhythmic drugs still rely on altering cardiac repolarization, they tend to target novel ion channel targets to avoid proarrhythmia, thereby increasing safety of therapy. Many sodium channel blockers, particularly slowly dissociating sodium channel blockers, or “class Ic drugs” according to the old Vaughan-Williams classification, suppress ectopy. In fact, this property was the initial goal for their development and their early, now abandoned, clinical use. An important, at times under-recognized, effect of such compounds is induction of refractoriness beyond the end of the action potential. This postrepolarization refractoriness appears to reduce inducible ventricular fibrillation and is a well-established effect of sodium channel blockers such as propafenone or flecainide4 but also of amiodarone and dronedarone.4–6 Amiodarone7,8 has served as a model compound for the development of new antiarrhythmic drugs, which are often analogues of amiodarone. Amiodarone is more effective than other clinically available antiarrhythmic drugs, and its proarrhythmic potential is relatively low.9 Therefore, it is the only antiarrhythmic agent that is recommended for use in patients with overt heart failure. This has resulted in the preclinical and clinical development of compounds such as SSR149744C, celivarone, ATI-2042, and dronedarone, among others, multichannel blockers that have similar chemical properties to amiodarone but contain less iodine and hence have less iodine-dependent unwanted effects.10,11 Celivarone has been tested for the suppression of defibrillator discharges in carriers of an implanted defibrillator but did not have an antiarrhythmic effect.12 The clinical development of celivarone has since been stopped. Dronedarone is an amiodarone analogue devoid of iodine. It is effective in the prevention of atrial fibrillation in patients with paroxysmal or persistent atrial fibrillation.13,14 The fact that dronedarone may be less effective in the prevention of atrial fibrillation than amiodarone in both indirect comparisons,13–15 and in a direct “head-to-head” comparison,16 suggests that other, potentially pharmacokinetic, factors favor amiodarone’s effectiveness (discussed next). Dronedarone was found to have a favorable safety profile in two large controlled trials that enrolled patients with nonpermanent, often recent-onset, atrial fibrillation.13,17 Indeed, the outcome of the ATHENA trial17 is the first signal ever recorded in clinical trials that rhythm control therapy with an antiarrhythmic drug can help to prevent outcomes such as hospitalization or cardiovascular death in patients with atrial fibrillation. Similar to amiodarone, dronedarone also has rate-controlling effects with a mean reduction in heart rate of 8 bpm.14,18 It has been speculated that this rate-controlling effect, or vascular/vasodilator effects of dronedarone, could have conveyed the benefit of dronedarone seen in the ATHENA trial. More recent data dispute this hypothesis: When given to patients with heart failure in the absence of atrial fibrillation19 or in permanent atrial fibrillation,20 dronedarone therapy causes harm, most likely as a result of unwanted, only partially understood effects of the drug in these populations. Hence, it remains likely that the positive outcome associated with dronedarone in the ATHENA trial (24% reduction in a composite of cardiovascular hospitalization or death, with significant reductions in cardiovascular death and in stroke)17 is the effect of a safe), moderately effective antiarrhythmic agent. Furthermore, at the time of this writing, there is a clear need to understand why reason dronedarone has detrimental effects in patients with heart failure. Azimilide is a potassium channel blocker that mainly inhibits IKr and is, similar to other potassium channel blockers, associated with torsades de pointes proarrhythmia.21 Its clinical antiarrhythmic efficacy appears to be relatively weak.7,8 A novel, experimentally available substance that can convert atrial fibrillation in experimental models is a selective inhibitor of the slow component of the delayed rectifier current, IKs (HMR1556).22 This substance does not interfere with the rapid component of the delayed rectifier current but nonetheless appears to bear proarrhythmic potential. This may point to the relevance of the total amount of available repolarizing currents for the risk for torsades de pointes (repolarization reserve23). Vernakalant is another multichannel blocker that is in clinical use for cardioversion of atrial fibrillation in Europe.24,25 Vernakalant preferentially prolongs the atrial action potential.26 The intravenous formulation rapidly terminates atrial fibrillation, which compares favorably with amiodarone.27 Vernakalant is recommended for cardioversion in the most recent atrial fibrillation guidelines in Europe.28 Without available data suggesting clinical ineffectiveness or safety concerns, the clinical development program of the oral formulation of vernakalant was stopped in the first half of 2011. To date, vernakalant is not approved for use in the United States. The outward currents that determine repolarization in the atria are slightly different than in the ventricle. In theory, inhibition of atrial-specific ion currents could be safer because such compounds would selectively prolong atrial action repolarization without affecting ventricular action potentials and QT interval (see Figure 112-2). One of the best-studied substances that attempts this approach is the relative selective inhibitor of the ultra-rapid component of the outward potassium current (IKur), AVE0118. AVE0118 prevents atrial fibrillation effectively in preclinical studies in goats,29,30 and restores atrial contractility after cardioversion.31 Despite these promising preclinical findings, there is no published record of antiarrhythmic efficacy in patients, and the company’s development of the oral compound has been stopped. Ranolazine, a multi-channel blocker that may predominantly inhibit the inward sodium current is approved for treatment of angina.32 Ranolazine may induce atrial postrepolarization refractoriness33 by inhibition of the late sodium current. Ranolazine reduced asymptomatic, nonsustained ventricular arrhythmias in a large study in postinfarction patients, albeit as a secondary outcome parameter.34 Preclinical data also suggest that ranolazine could prevent torsades de pointes, at least in models of the long-QT syndrome.13,14 The clinical antiarrhythmic effects of ranolazine have not been systematically studied in prospective trials thus far. Several medium-sized clinical trials are under way to assess the clinical efficacy of ranolazine to prevent atrial fibrillation and to prevent defibrillator shocks. Ivabradine is a new type of ion channel–blocking drug that is in clinical use as an antianginal agent. Ivabradine inhibits the “funny current,” a hyperpolarization-activated inward current that is believed to drive impulse generation in the sinus node.29 This current, most likely generated by ion flow through proteins coded by the HCN gene family, is an attractive drug target because the target protein isoform is predominantly expressed in sinus node and in retina. Consequently, side effects are rare and mainly limited to intermittent flashlike visual sensations secondary to interactions of the drug with retinal channels. Although these occur in 12% of treated patients, they are rarely therapy-limiting. Ivabradine has been approved for the treatment of patients with angina pectoris who have contraindications for β-adrenoreceptor blockade35,36 and can be used safely in patients with heart failure.36a The mode of action strongly suggests that ivabradine may be a specific and effective treatment for patients with inadequate sinus tachycardias, and several patient series report a favorable effect of ivabradine in such patients.37–39 Consistent with its mode of action, ivabradine may hence be helpful in the management of selected patients with inappropriate sinus tachycardias. Potassium channel openers such as nicorandil or NS1643 (although they mainly activate ATP-sensitive potassium channels) may be able to reverse the action potential prolongation associated with drug-induced proarrhythmia and arrhythmias in the long-QT syndromes.40,41 Such substances could theoretically help to acutely treat proarrhythmic situations associated with prolongation of the ventricular action potential. This may apply to patients with congenital long-QT syndromes but also to patients with acquired forms of torsades de pointes. An antiarrhythmic effect has been reported in case reports42 but has not been tested in larger trials. Dietary supplements containing fish oil or omega-3 fatty acids have been proposed as novel, safe antiarrhythmic agents,43 with some reports suggesting prevention of recurrent atrial fibrillation44–46 and others not finding any effects on atrial fibrillation,47 including prospective controlled trials of postoperative atrial fibrillation48,49 (and a large trial of spontaneous atrial fibrillation recurrence).49 Conflicting results could be a result of differing dosage and length of treatment45 but may also reflect publication bias, which would favor positive effects. Biologically, fatty acids could have direct effects on sarcolemmal ion channels50 or mediate modification of the cardiac action potential through alterations in high-density lipoprotein cholesterol.51 Overall, their antiarrhythmic effects appear to be weak. It is assumed that abnormal calcium release within the cardiomyocyte may provoke triggered activity promoting drug-induced ventricular proarrhythmia. A fix combination of verapamil and quinidine, another form of a multichannel blocker that has been used for many years in Germany, is as effective as sotalol for the prevention of atrial fibrillation but appears to provoke less proarrhythmia than quinidine alone: In the PAFAC and SOPAT trials, telemetric ECG monitoring detected one ventricular tachycardia in 895 patients treated with the quinidine-verapamil combination over one year compared with 8 episodes of torsades de pointes in 645 patients treated with sotalol over one year.52,53 Another fix combination that is in clinical development is a fix combination of dronedarone and ranolazine, which will have a stronger inhibitory effect on the late sodium current than dronedarone alone and add the multichannel-blocking properties of dronedarone to ranolazine monotherapy. Two inward rectifying potassium currents, the background current IK1 and the acetylcholine-activated current IK,ACh, are altered in atrial fibrillation, with IK1 being increased and IK,ACh becoming in part constitutively active.37,54 As a consequence, the refractory period is shortened. This creates a condition that supports multiple wavelet reentry and periodic activity of sustained, high-frequency functional reentry sources described as “rotors.”55,56 In addition, a shift of the resting membrane potential toward more positive potentials via atypical potassium channels or unselective membrane pores may increase the chance that spontaneous depolarizations (e.g., afterdepolarizations) can activate sodium channels and thereby initiate a premature response. Restoration of normal function of these currents appears to be an attractive target for antiarrhythmic drug therapy, especially in atrial fibrillation. Inhibition of stretch-activated channels can prevent atrial fibrillation in experimental models.57 Although attractive, inhibitors of stretch-activated channels require more research before they may reach clinical applicability, because the only available specific inhibitor of stretch-activated channels is a peptide with all the pharmacodynamic difficulties inherent to such compounds. Abnormal regulation of intracellular calcium can be found in fibrillating human atria.58,59 There is a growing body of evidence to suggest that both atrial ectopy and electrical remodeling are highly calcium-dependent phenomena. Stabilization of abnormal calcium release from the sarcoplasmic reticulum can prevent ventricular tachyarrhythmias in genetically modified models of altered calcium release.60–63 Whether the calcium-stabilizing interventions that have been investigated in the context of ventricular arrhythmias can also provide antiarrhythmic action in atrial fibrillation remains to be tested. Contrary to the origin of many episodes of atrial fibrillation that may have a focal origin at the junction of the pulmonary veins and in the posterior left atrium, initiation of ventricular fibrillation is often a consequence of several synergistic and regulatory processes that combine to provoke triggered activity that may occur in any part of the ventricular myocardium.64 Several observations have long suggested a relevant role for calcium in sudden death: Prolongation of the ventricular action potential in hypertrophied ventricles and pause-dependence of ventricular fibrillation suggest that afterdepolarizations may be relevant for sudden death. Autonomic imbalance, and specifically excessive sympathetic stimulation, may itself also provoke ventricular arrhythmias, most likely by increased intracellular calcium levels in subcellular microdomains and facilitated calcium release from the sarcoplasmic reticulum. Furthermore, it is well known that diastolic calcium levels are increased in heart failure, whereas systolic calcium release is increased in hypertrophy. Consequently, abnormally increased systolic calcium release, at times combined with increased diastolic calcium levels, provokes ventricular fibrillation in models of cardiac hypertrophy65,66 and may contribute to ventricular arrhythmias in other forms of cardiac hypertrophy and failure.67 Abnormal diastolic calcium release from the sarcoplasmic reticulum underlies catecholaminergic ventricular tachycardias in transgenic models.61,62 Prolonged calcium release from the contractile apparatus of cardiomyocytes may have similar effects in some forms of “hypertrophic” cardiomyopathy.68 Calcium channel blockade by existing drugs (verapamil or, with a more specific action on the sarcoplasmic reticulum calcium release channels, flecainide)69 may in selected patients suppress ventricular arrhythmias, e.g., suffering from catecholaminergic ventricular tachycardia.69,70 Ranolazine may also be able to suppress calcium release, at least in experimental settings.71 Experimental evidence suggests that early afterdepolarizations often occur secondary to abnormal activity of the sodium-calcium exchanger.72 Hence, inhibitors of the sodium-calcium exchanger are under preclinical evaluation for trigger suppression, but their antiarrhythmic efficacy has not yet been proven.73,74 Inhibitors of intracellular kinases (e.g., CaMKII inhibitors61,66,75,76), and possibly also protein phosphatase inhibitors, can acutely suppress ventricular arrhythmias in a variety of experimental models (e.g., hypertrophy, infarction, electrical storm). Inhibition of spontaneous diastolic calcium leaks from the sarcoplasmic reticulum (e.g., activation of FKBP 12.6 by JTV519 and related substances)61 can suppress ventricular arrhythmias associated with genetic modifications, causing leaks in the sarcoplasmic reticulum,58,77,78 and probably also in models with reduced calsequestrin function79 or increased triadin function.62,71,72 It is possible that store-operated calcium also contributes to calcium release–dependent arrhythmogenesis, either indirectly via inducing cardiac hypertrophy80 or directly.74,75 A thorough understanding of the subcellular calcium compartments that undergo independent calcium regulation may help to devise new compounds that inhibit arrhythmogenesis while maintaining the essential effects of calcium on contractile function, regulation of protein function, and regulation of gene expression.
New Antiarrhythmic Drugs and New Concepts for Old Drugs
Background and Current Clinical Context
Main Effects of Antiarrhythmic Drugs
Prolongation of Repolarization
Induction of Postrepolarization Refractoriness
New Antiarrhythmic Drugs
Fish Oil and Omega-3 Fatty Acids
Combining Antiarrhythmic Drugs in Fix Combinations
New Targets for Antiarrhythmic Drugs
Ion Channel Targets
Abnormal Calcium Release and Arrhythmia Initiation
New Antiarrhythmic Drugs and New Concepts for Old Drugs