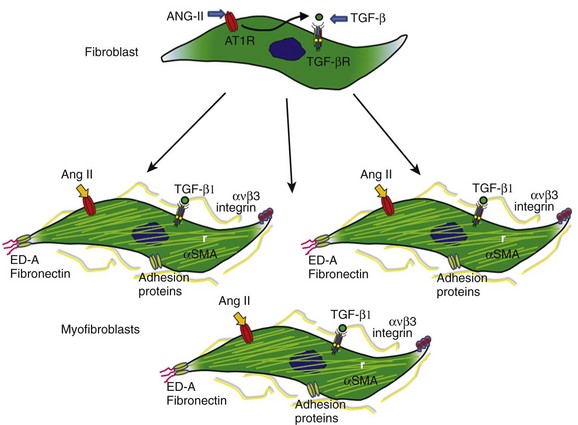
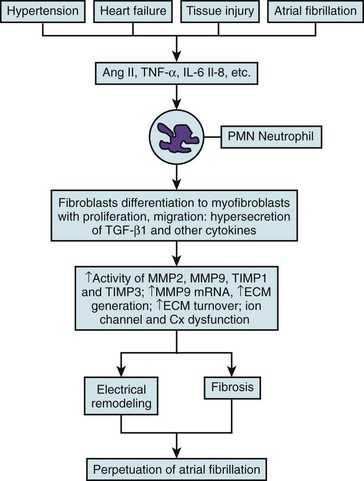
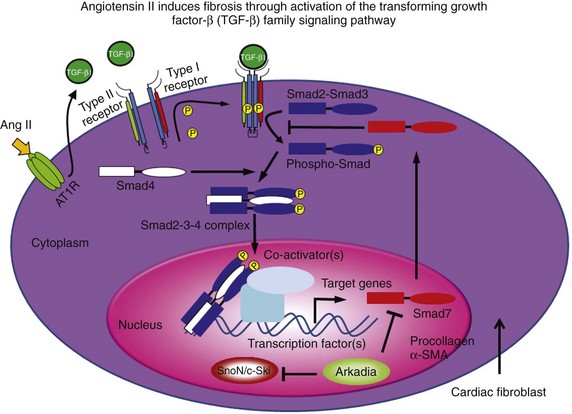
46 Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia seen in clinical practice. It is the most important cause of embolic stroke and is associated with increased morbidity and mortality.1 Yet despite more than 100 years of basic and clinical research, we still do not fully understand its fundamental mechanisms, and we have not learned how to treat it effectively. When AF lasts continuously for more than 7 days, it is designated as persistent AF.2 Spontaneous, pharmacologic, or ablative resumption of sinus rhythm is infrequent in persistent AF, with prompt recurrences or commonly failed cardioversions. Episodes lasting longer than 1 year are termed “long-term persistent AF.” As to the causes of persistent AF, it is reasonable to speculate that continuous high-frequency and irregular bombardment of the atrial cells and tissues with fibrillatory waves during long-lasting AF leads to a modification of the molecular substrate on which waves propagate, with consequent electrical and structural remodeling, substantial enough to increase the likelihood of perpetuation of the electrical sources that maintain the arrhythmia. Fibrosis has been implicated in the initiation and maintenance of arrhythmia, affecting electrical propagation through slow, discontinuous conduction with “zigzag” propagation,3,4 reduced regional coupling,5 abrupt changes in fibrotic bundle size,6 and micro-anatomical reentry.7 In the past, most clinical, experimental, and numeric studies regarded fibrosis as electrically insulating obstacles. However, heart injury promotes fibroblast differentiation into myofibroblasts,8,9 which are hypercontractile and hypersecretory of soluble proteins termed cytokines, known to affect myocardial function, and have been shown to electrotonically couple with myocytes in vitro.10–14 Thus, it is likely that both fibroblasts and fibrosis play critical roles in atrial electroanatomical remodeling and further facilitate perpetuation of AF. Yet AF per se may promote cardiac fibrosis, and it is notable that although numerous studies point to a clear association between AF and fibrosis, the underlying pathologic basis for AF-induced fibrosis remains poorly understood. Arguably, early identification and prevention of the molecular and cellular mechanisms of the atrial fibrotic process may halt the progression of AF and potentially improve response to both pharmacologic and nonpharmacologic therapy. This highlights the importance of investigating the biology of the fibroblasts as “sentinel cells”15; their role in the fibrotic process; the impact of their interactions with myocytes; their effects on sarcolemmal ion channel behavior, cardiac excitability, and atrial impulse propagation; and their possible role in the mechanisms underlying the transition from paroxysmal to persistent AF, as well as stabilization of the latter. In this chapter, we present a brief and necessarily incomplete review of possible mechanisms underlying AF-induced fibrosis. We pay particular attention to the role of activated fibroblasts (myofibroblasts) and the cytokines released by them in the mechanisms of electrical and structural remodeling. Our objective is twofold: first, to go over our understanding of the molecular mechanisms of fibrosis; and second, to help predict how myofibroblasts impinge on the electrical function of atrial myocytes by means of signaling molecules, resulting in the electrophysiological remodeling that contributes to AF perpetuation. Cardiac fibroblasts are critical players in normal cardiac function.8 Although they occupy a small portion of the myocardial tissue volume, cardiac fibroblasts account for 50% to 70% of the cells of the normal adult mammalian heart,9 and even a greater proportion in pathologic conditions where cardiac fibrosis ensues as a result of the transformation of fibroblasts to myofibroblasts, which is a key event in connective tissue remodeling (Figure 46-1).8,10,11 Cardiac myofibroblast proliferation and migration with concomitant collagenous matrix accumulation leading to fibrosis develops during myocardial remodeling in ischemic, hypertensive, hypertrophic, dilated cardiomyopathies,12 arrhythmogenic right ventricular cardiomyopathy (ARVC),13 and heart failure. Similarly, myofibroblasts are active contributors to atrial fibrosis,8 which is part of the maladaptive atrial response to AF.14 Myofibroblasts are phenotypically transformed (active) fibroblasts that contain α-smooth muscle actin and express integrins, fibronectin, and other adhesion proteins (see Figure 46-1). They were first identified by Gabbiani and Majno, who gave them the name myofibroblasts15 after demonstrating their contractility using chemical mediators of inflammation and strips of granulation tissue. These wound-healing fibroblast-like cells are involved in each of the fibrous tissue responses present throughout the body. Yet their role in the mechanisms underlying structural and electrophysiological remodeling secondary to sustained atrial fibrillation has not been fully elucidated. Much of the available information on the specific role played by myofibroblasts in atrial fibrosis and AF perpetuation is derived from what is known about hypertensive heart disease, in which impaired tissue compliance and symptomatic failure are linked to interstitial fibrosis and the expression of extracellular matrix genes, including type I and III fibrillar collagens and fibronectin. It has been shown that fibroblast proliferation and collagen deposition are significantly and heterogeneously increased in all atrial regions after prolonged AF,16,17 and that the development of atrial fibrosis is highly controlled by the renin-angiotensin-aldosterone system and by downstream activation of soluble proteins termed cytokines, particularly transforming growth factor-beta1 (TGF-β1) and platelet-derived growth factor (PDGF). Figure 46-1 The phenotypic transformation (activation) of fibroblasts into myofibroblasts is highly controlled by the renin-angiotensin-aldosterone system and by downstream activation of soluble proteins termed cytokines, particularly transforming growth factor beta-1 (TGF-β1). Myofibroblasts proliferate and hypersecrete α-smooth muscle actin (αSMA), which makes them hypercontractile and promotes their expression of integrins and other adhesion proteins, as well as extracellular matrix genes, including fibronectin type I and type III fibrillar collagens. To fully understand the role of myofibroblasts and/or cytokines in fibrosis and AF perpetuation, it is necessary to fully understand the role played by inflammation, which is known to play an important pathogenic role leading to fibrosis in several cardiovascular diseases.18 Increasing evidence now suggests that inflammatory pathways are directly linked to cellular and subcellular mechanisms known to lead also to AF.18 Activated inflammatory cells (such as neutrophils, lymphocytes, monocytes, and resident macrophages), proinflammatory cytokines, and activated platelets are important players in this picture (Figure 46-2). Moreover, inflammatory cascades alter ion channel function in myocytes and are strongly associated with fibrosis.18 Inflammatory stimuli such as nicotine adenine dinucleotide phosphate (NADPH)-oxidase–derived reactive oxygen species (ROS), cytokines, growth factors, angiotensin-II (Ang-II), and other hormones, as well as mechanical stretch, are well-known triggers of fibroblast proliferation, migration, and differentiation into myofibroblasts, which are critical players in the development of fibrosis.18 Figure 46-2 Inflammatory pathways involved in the perpetuation of atrial fibrillation. Hypertension, heart failure, tissue injury, or sustained atrial fibrillation (AF) leads to the release of proinflammatory cytokines and hormones, such as angiotensin-II, tumor necrosis factor (TNF)-α, and interleukin-6 and interleukin-8, which promote activation of leukocytes with subsequent activation of fibroblasts into myofibroblasts, which proliferate and become hypersecretory of a number of cytokines. As a result, connexin and ion channel dysfunction occurs, along with myocyte apoptosis and matrix generation and turnover, which lead to electrical and structural remodeling (fibrosis) that contributes to the perpetuation of AF. The concept that AF promotes inflammation was derived from studies demonstrating that chronic human AF is associated with increased atrial oxidative stress and peroxynitrite formation.19 Thereafter, long-term rapid atrial pacing experiments in dogs demonstrated pacing-induced shortening of the atrial effective refractory period, which was associated with decreased tissue ascorbate levels and increased protein nitration. The latter is a biomarker of peroxynitrite formation.20 Oral ascorbate supplementation attenuated all changes. In a parallel study, supplemental ascorbate was given to 43 patients before, and for 5 days after, cardiac bypass graft surgery.20 Patients receiving ascorbate had a 16.3% incidence of postoperative AF compared with 34.9% in control subjects. More recently, rapid atrial pacing in a porcine model increased the activity of the profibrotic enzymes matrix metalloproteinase (MMP)2 and MMP9 in the atrial interstitium, increased MMP9 mRNA expression, and increased the activity of tissue inhibitor of matrix metalloproteinase (TIMP)1 and TIMP3.21 Figure 46-2 illustrates a likely scenario linking sustained AF to inflammation with subsequent electrical remodeling, fibrosis, and atrial fibrillation perpetuation. Proinflammatory cytokines and hormones, such as Ang-II, tumor necrosis factor-α (TNF-α), interleukin (IL)-6, and IL-8, related to cardiovascular disease and tissue injury, promote activation of leukocytes with subsequent release of cytokines and ROS. As a result, connexin and ion channel dysfunction occur, along with myocyte apoptosis and matrix generation and turnover, which leads to electrical and structural remodeling that predisposes an individual to AF.18 Clinical studies have identified inflammation as a pathogenic factor in AF, as well as a correlation between inflammatory markers and the incidence of AF.22 In a 5-year follow-up of an original Framingham Heart Study cohort of about 800 patients with no history of AF, subjects’ white blood cell count was significantly associated with AF development after correction for standard risk factors for AF.23 As a standard indicator of general inflammation, white blood cell count is also an indicator of the role of systemic inflammation as a risk factor for AF. Inflammatory markers appear to be good indicators of failure to terminate AF after cardioversion using both pharmacologic and electrical methods. Inflammatory markers such as serum amyloid A (SAA) and C-reactive protein (CRP) were studied before and 3 weeks after cardioversion in patients with persistent AF24; 33% of patients presented with AF recurrence at 3 weeks after cardioversion, and levels of CRP and SAA were associated with recurrence.24 Patients with persistent AF had a higher inflammatory index than patients with proximal AF, as measured by serum CRP levels, which suggested progressive inflammation in association with AF duration/severity.25 Patients with a history of AF have been shown to have increased inflammatory CD45+ cell infiltration in the atrial myocardium compared with patients with no AF.26 Further support for the idea that inflammatory mediators play a significant role in AF comes from studies demonstrating that hydrocortisone treatment before cardiac surgery reduced the incidence of postoperative AF.27 The development of interstitial fibrosis and its impact on atrial contractile and electrical function have been found to be central for AF maintenance.17,28,29 Fibroblast proliferation and collagen deposition are significantly and heterogeneously increased in all atrial regions after prolonged AF,16,17 a process that is highly controlled by the renin-angiotensin-aldosterone system, and by downstream activation of soluble cytokines such as TGF-β1 and PDGF. Originally termed cytokines by Stanley Cohen in 1974, these proteins are major players in immune reactions.30 However, although immune cells are a major source of cytokines, a number of other cell types, including heart cells, can synthesize and release them.31 Multiple studies have demonstrated close relationships between elevated cytokines and AF. In patients with paroxysmal or persistent AF, serum levels of vascular endothelial growth factor (VEGF), IL-8, soluble intercellular adhesion molecule 1 (sICAM-1), and TGF-β1 are upregulated at baseline,32 and increased VEGF has been shown to associate independently with AF occurrence.33 Levels of IL-18 are also higher in patients with persistent AF with atrial dilatation.34 Graded increases in TNF-α and N-terminal pro-brain natriuretic peptide (NTpBNP) have been shown in various AF groups, and have been highest in permanent AF.35 A cross-sectional analysis of the Heart and Soul Study patients suggested a genetic link between IL-6 polymorphism 174CC and AF. Patients with this polymorphism have high levels of IL-6.36 Increased serum levels of cytokines and other inflammatory markers correlate also with failure of rhythm control in AF.37 For example, it has been shown that expression of IL-2 is associated with the outcome of cardioversion with amiodarone, and serum levels of IL-2 were lower in patients who cardioverted successfully compared with those in noncardioverted patients. In addition, the baseline level of IL-2 was a strong indicator of therapeutic success.37 In patients with paroxysmal or persistent AF, elevated plasma levels of IL-6 and CRP appear to be independent predictors of failure of pulmonary vein isolation.38 In another study, baseline levels of CRP, TNF-α, sICAM-1, and oxidative stress markers were elevated in patients with AF compared with controls, and were higher in patients with than in those without persistent AF recurrence; however, IL-6 levels were equally elevated in the two subgroups.39 In yet another clinical study, serum concentrations of IL-6, CRP, and CD40L decreased in patients with no AF recurrence after ablation, whereas the same indicators did not decrease in patients who failed ablation.40 An AF-related release of cytokines was suggested by another study, in which levels of CRP and IL-6 were similar in AF and sinus rhythm (SR) patients when in SR. In AF patients, single episodes of AF led to these increased protein levels.41 Along with increases in the proinflammatory proteins, AF is associated with decreases in anti-inflammatory mediators. Peroxisome proliferator–activated receptor-γ (PPARγ), a multifunctional nuclear receptor protein, regulates a number of transcription factors, and has been shown to be anti-inflammatory.42 mRNA and protein levels of PPARγ were shown to be decreased in hypertensive patients with AF compared with hypertensive patients with no AF and were further decreased in persistent versus proximal AF patients. In addition, a negative correlation was noted between levels of inflammatory cytokines and PPARγ.43 Members of the TGF-β family of pleiotropic cytokines are implicated in a wide variety of cell functions.44 They regulate inflammation, extracellular matrix deposition, and cell proliferation, differentiation, and growth. In mammalian species, three structurally similar isoforms of TGF-β (TGF-β1, TGF-β2, and TGF-β3), encoded by three distinct genes, have been identified.45 The most prevalent is TGF-β1. It expresses widely in many cell types, whereas the other isoforms are found in a more limited range of cells and tissues.44 Binding of TGF-β family ligands induces the association of type II and type I receptors into a heterodimeric complex (Figure 46-3). The type II receptor kinase phosphorylates the type I receptor, inducing its serine/threonine kinase activity. Receptor-regulated Smads (R-Smads) are then activated by phosphorylation by the type I receptor kinase. Activated R-Smads form complexes with common-partner Smads (Co-Smad), and translocate into the nucleus.14 Once there, these proteins bind other transcriptional factors, including both transcriptional coactivators and corepressors. Figure 46-3 Angiotensin-II (Ang-II) induces fibrosis through activation of the transforming growth factor (TGF)-β–signaling pathway in the myofibroblast. Activation of the angiotensin type 1 receptor (AT-1R) increases the expression of TGF-β1, which leads to phosphorylation of type I and type II TGF-β1 membrane receptors. This leads to the formation of a complex that activates Smad2-Smad3 through phosphorylation, promoting consolidation of the Smad2-3-4 complex with subsequent translocation to the nucleus, leading to increased transcription of pro-fibrillatory genes. Ang-II/AT-1R-specific activation of Arkadia-mediated poly-ubiquitination and degradation of Smad7 are thought to decrease the inhibitory feedback regulation of TGF-β1/Smad signaling and serve as key mechanisms for AF-induced atrial fibrosis.14 Knowledge about the effects of TGF-β1 in advancing cardiac hypertrophy and fibrosis in vivo derives in part from studies in transgenic mice. TGF-β1–overexpressing mice demonstrated significant cardiac hypertrophy accompanied by interstitial fibrosis.46 On the other hand, cardiac-restricted expression of a mutant TGF-β1 that enhances local TGF-β activity was associated with atrial but not ventricular fibrosis,47 which suggested that the susceptibility of the atrial myocardium to the fibrogenic actions of TGF-β is greater than that of the ventricular myocardium. Fibrotic remodeling of the atrium in this transgenic model was sufficient to increase vulnerability to AF.48 A single-nucleotide polymorphism at codon 25, 915 G-> C, which is associated with myocardial infarction and atrial stiffness, is also associated with increased plasma TGF-β1 levels in patients with essential hypertension and AF.49 Lower atrial voltage, especially in the left atrium, has been associated with AF. Plasma levels of TGF-β1 have been shown to correlate strongly with left atrial voltage and volume, suggesting that TGF-β1 is an indicator of AF.50 In an interesting study in which right atrial appendages were taken from SR and AF patients who were divided into five groups (SR; paroxysmal/chronic persistent AF of <6 months; and chronic permanent AF of 7 to 24 months, 25 to 60 months, and >60 months), a gradual increase in fibrosis was noted as AF progressed.51 Similar to collagen, progressive increases in the expression of TGF-β1 mRNA, protein, and TGF-β receptor II protein were noted. However, the TGF-β1
Myofibroblasts, Cytokines, and Persistent Atrial Fibrillation
Cardiac Fibroblasts and Myofibroblasts
Inflammation and AF
Cytokines and Human AF
TGF-β1 and AF
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Myofibroblasts, Cytokines, and Persistent Atrial Fibrillation
