Myocarditis and Specific Cardiomyopathies: Introduction
Inflammatory cardiomyopathies, particularly viral myocarditis, have served as models to understand the development of heart failure. More than 70 different specific cardiomyopathies associated with general systemic disease, neuromuscular disorders, hypersensitivity and toxic reactions, and the peripartum state have been described. A list of causes associated with the development of cardiomyopathy is presented in Fig. 35–1. When they are considered as a group, these disorders are infrequent; considered individually, they are rare. Cardiomyopathy associated with HIV disease is considered in Chap. 93.

Myocarditis
In its most literal sense, myocarditis means inflammation of the myocardium. As early as 1806, a relationship between infection (diphtheria) and chronic heart disease was postulated, but it was not until the 1970s, with the advent of endomyocardial biopsy, that the diagnosis of myocarditis could be established during life. Multiple infectious etiologies (Table 35–1)1 have been implicated as the cause of myocarditis, the most common being viral, specifically, the enterovirus coxsackie B. In the majority of patients, active myocarditis remains unsuspected because the cardiac dysfunction is subclinical and self-limited. The discovery of myocarditis in 1% to 9% of routine postmortem examinations suggests that myocarditis is a major cause of sudden, unexpected death.2
| Infectious |
| Viruses: Coxsackievirus, echovirus, HIV, Epstein-Barr virus, influenza, cytomegalovirus, adenovirus, hepatitis A and B, mumps, poliovirus, rabies, |
| respiratory syncytial virus, rubella, vaccinia, varicella zoster, arbovirus |
| Bacteria: Corynebacterium diphtheriae, Streptococcus pyogenes, Staphylococcus aureus, Haemophilus pneumoniae, Salmonella spp., Neisseria gonorrhoeae, Leptospira spp., Borrelia burgdorferi, Treponema pallidum, Brucella spp., Mycobacterium tuberculosis, Actinomyces spp., Chlamydia spp., Coxiella brunetti, Mycoplasma pneumoniae, Rickettsia spp |
| Fungi: Candida spp., Aspergillus spp., Histoplasma spp., Blastomyces spp., Cryptococcus spp., Coccidioidomycosis spp. |
| Parasites: Trypanosoma cruzii,Toxoplasma spp., Schistosoma spp. Trichina spp. |
| Noninfectious |
| Drugs causing hypersensitivity reactions |
| Antibiotics: Sulfonamides, penicillins, chloramphenicol, amphotericin B, tetracycline, streptomycin |
| Antituberculous: Isoniazid, para-aminosalicylic acid |
| Anticonvulsants: Phenindione, phenytoin, carbamazepine |
| Anti-inflammatories: Indomethacin, phenylbutazone |
| Diuretics: Acetazolamide, chlorthalidone, hydrochlorothiazide, spironolactone |
| Others: Amitriptyline, methyldopa, sulfonylureas |
| Drugs not causing hypersensitivity reactions |
| Cocaine, cyclophosphamide, lithium, interferon-α |
| Nondrug causes |
| Radiation, giant cell myocarditis |
Infection by cardiotropic viruses prompted the initial hypothesis that the viral infection was responsible for myocardial injury. However, several investigators noted that cardiac dysfunction increased after the eradication of the infective agent and speculated that the pathogenesis of myocarditis can be caused by two distinct phases of myocardial cell damage—the first caused by direct viral infection and the second caused by the host’s immune response (Fig. 35–2). Support for this theory comes initially from the work of Woodruff,3 who noted that the histologic evidence of cardiac injury in coxsackie B infection appeared only after the virus was no longer detectable in the myocardium. Subsequently, demonstration of T lymphocyte and macrophage infiltration, perforin granules, and a variety of cytokines known to depress myocardial contractility in endomyocardial biopsies of patients with active carditis strengthened the concept of immune-mediated injury.
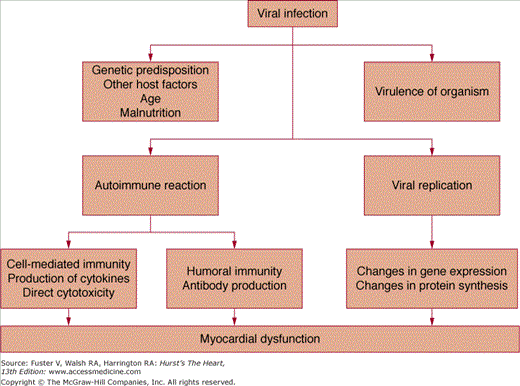
Our understanding of the specific immune responses that lead to myocardial injury is derived largely from animal models of myocarditis induced by cardiotropic viruses.4 A timeline for experimental viral myocarditis is shown in Fig. 35–3. After gaining entry to the body from the gastrointestinal tract (enterovirus) or through the respiratory tract (adenovirus and enterovirus), these cardiotropic viruses bind to the coxsackie-adenoviral receptor (CAR), which in turn allows for the incorporation of the viral genome into the myocyte.5 In the acute phase of viral myocarditis (days 0-3), mice injected with coxsackievirus show evidence of direct viral cytotoxicity, with myocyte necrosis in the absence of an inflammatory cell infiltrate. Activated macrophages begin to express interleukin (IL)-1, IL-2, tumor necrosis factor α (TNF-α), and interferon-γ (IFN-γ).6
Figure 35–3
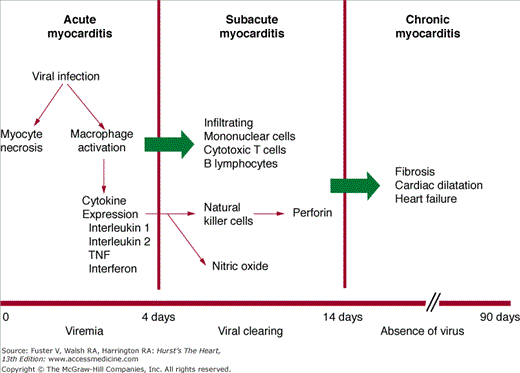
Timeline of viral myocarditis. TNF, tumor necrosis factor. Reproduced with permission from Kawai.4
This ushers in the second or subacute phase (days 4–14) of myocarditis, which is characterized by the infiltration of natural killer (NK) cells, production of neutralizing antibody, and cell-mediated immunity. The first wave of infiltrating cells consists mainly of NK cells that limit viral replication by destroying virus-infected cells. NK cells release perforin and granzymes, which form circular pore lesions on the membrane surface of virus-infected cells.7 Uninfected cells are spared from injury. Antibody production follows viral replication. After invading the myocardium, coxsackievirus increases its expression, becoming maximal on day 4. Almost no neutralizing antibody is present until day 4, after which titers quickly rise, becoming maximal on day 14. The increase in antibody titer is closely related to the elimination of virus from the heart. Infiltrating mononuclear cells appear in the myocardium between 5 and 10 days after viral infection.3 T lymphocytes are attracted to the myocardium through classic cell-mediated immunity. Within the cytoplasm of infected myocytes, viral particles are broken down into peptide fragments and then placed on the cell surface in association with major histocompatibility complex class 1 antigen. Through their T-cell receptor (TCR), cytotoxic T-cells recognize virus-infected cells and destroy them by either cytokine production or perforin-mediated cell cytolysis.4 In combination with neutralizing antibodies, these mononuclear cells help to suppress viral infection and limit cardiomyocyte damage. When host defense mechanisms are inadequate, chronic viral infection produces widespread cardiac damage and eventual cardiac dilatation and failure.8 Conversely, continuous exuberant T-cell activation can be equally destructive. This occurs when antigens intrinsic to the myocardium cross-react with viral peptides, leading to sustained T-cell activation (so-called molecular mimicry), which can ultimately lead to dilated cardiomyopathy (DCM).9
Cytokines are a major mediator of immune activation and its maintenance. Circulating levels of IL-1, IL-2, and IL-6 are elevated in patients with acute myocarditis, as are TNF-mRNA and protein expression. In animal models, overexpression of TNF-α produces florid myocarditis,10 but mice deficient in the TNF p55 receptor (TNF-R1–/–) experience milder autoimmune myocarditis.11 However, a recent study by Wada and colleagues12 suggests that TNF-α can actually play a protective role in the acute stage of viral myocarditis. The observed full recovery of some patients with severe left ventricular (LV) dysfunction may be the result of short-term exposure to these inflammatory cytokines.9
A third or chronic phase of viral myocarditis (days 15-90) occurs after the elimination of virus and is typified by insidious ongoing myocardial damage.4 Hearts from infected mice are hypertrophied, and myocardial fibrosis is prominent. Inflammatory cells are no longer seen. The mechanisms involved in the transition from this stage to the development of DCM are not fully understood, but three mechanisms seem likely—persistent viral infection, ongoing autoimmune destruction, and apoptosis.
Attempts to culture virus from human myocardial tissue have generally been unsuccessful. However, identification of viral genomic fragments in myocardial samples by in situ hybridization and polymerase chain reaction from patients with myocarditis and DCM has been reported.13 These genomic fragments may not be capable of replicating as intact cardiotropic virus but probably serve as a persistent source of antigen to drive the deleterious immune responses. Whereas persistence of viral genomes in the myocardium of patients with LV dysfunction has been associated with a progressive impairment of ventricular function, spontaneous resolution has been associated with improvement in the LV ejection fraction (LVEF).14
Autoimmune-mediated heart disease results from a process of molecular mimicry whereby antibodies to viral proteins cross-react with structural elements of the heart. For example, injection of myosin into susceptible mice produces active myocarditis.15 One reason for this may lie in the fact that myosin shares approximately 40% identity with the amino acid sequence of Coxsackie virus CVB3 capsid protein. Several other circulating heart-specific autoantibodies have been identified, including antisarcolemma antibody, which also exhibits cross-reactivity with CVB3 capsid protein.16 Further investigation with this virus-free model of myocarditis will help in elucidating the pathogenesis of molecular mimicry.
Apoptosis, or programmed cell death, may be a third pathogenic mechanism leading from myocarditis to DCM. Although this association is currently speculative, certain viruses have been identified as triggers of apoptosis.17 Cytokines can activate death-domain sequences or ceramide-mediated signaling pathways as part of the remodeling process.18 Although animal models of myocarditis may progress to DCM, as can patients with clinically suspected or biopsy-proven myocarditis, the percentage of patients with idiopathic DCM representing the end stage of an active myocarditis is unknown.
The clinical manifestations of myocarditis are variable, ranging from an asymptomatic or self-limited disease in some to profound cardiogenic shock in others. Cardiac involvement typically occurs 7 to 10 days after a systemic illness. The most obvious symptom suggesting myocarditis is an antecedent viral syndrome with fever, myalgias, and malaise. The majority of patients have no specific cardiovascular complaints but can have ST-segment and T-wave abnormalities on the ECG suggesting myocarditis. Chest pain may occur in up to 35% of patients and may be typically ischemic, somewhat atypical, or pericardial in character. Chest pain usually reflects associated pericarditis but occasionally may result from myocardial ischemia.19
Acute DCM from lymphocytic myocarditis may produce mild, moderate, or fulminant heart failure. The vast majority of patients with mild symptoms have spontaneous recovery in ventricular function and normalization of heart size. Patients with New York Heart Association (NYHA) class III or IV heart failure typically have greater degrees of ventricular dilatation and dysfunction. Although some of these patients recover spontaneously, it is estimated that 50% of them will be left with residual myocardial dysfunction, and 25% will either die or require cardiac transplantation.19 Biopsy-proven relapses have occurred in some patients, and recurrent myocarditis should be suspected if ventricular function subsequently deteriorates.20 Fulminant myocarditis is often dramatic and accompanied by a rapid onset of symptoms.21 Patients are severely ill, with circulatory collapse and evidence of end-organ dysfunction. They frequently have fever, severe global myocardial dysfunction, and a minimal increase in LV end-diastolic dimension. Mechanical circulatory support can be required, as a bridge to either cardiac transplantation or recovery.
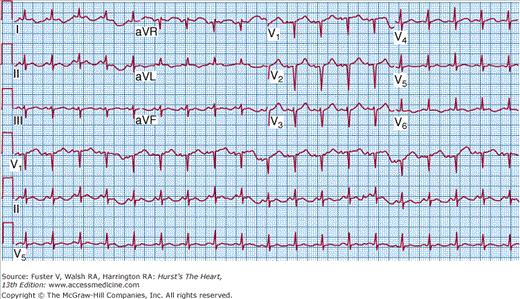
Occasionally, patients present with a clinical syndrome identical to acute myocardial infarction, with ischemic chest pain and ST-segment elevations on the electrocardiogram (ECG) (Fig. 35–4). LV dysfunction, when present, tends to be diffuse rather than segmental. At autopsy, the coronary arteries are usually widely patent, although viral coronary arteritis has been reported.22 Coronary vasospasm has also been associated with acute myocarditis.
Patients may present with syncope or palpitations from atrioventricular (AV) block or ventricular arrhythmia. Complete AV block is common, with some patients presenting with Stokes-Adams attacks. Sudden cardiac death is the initial presentation of myocarditis in some patients, presumably from complete heart block or ventricular tachycardia. In a 25-year review of sudden death among military recruits, 20% had myocarditis documented at autopsy.23 In some patients with refractory ventricular arrhythmias, endomyocardial biopsy or autopsy has revealed myocarditis.
Endomyocardial biopsy is the critical test to establish a diagnosis of myocarditis. Endomyocardial biopsy techniques enable the repetitive sampling of the human myocardium with minimal discomfort, minor morbidity, and a mortality rate of 0.2%.24 Right ventricular (RV) myocardial specimens can be obtained by accessing the right internal jugular or femoral vein. Biopsy of the LV is infrequently performed because of the higher morbidity associated with this approach. The RV bioptome is positioned under fluoroscopy or echocardiography to sample the interventricular septum. Because myocarditis can be focal, a minimum of four to six fragments is obtained. Endomyocardial biopsy must be applied as quickly as possible to maximize the diagnostic yield. Resolution of active myocarditis has been documented within 4 days of initial biopsy, with progressive clearing over several weeks on serial biopsy.25 Diagnoses that can be made or confirmed by endomyocardial biopsy are listed in Table 35–2.
| 1. Myocarditis |
| Giant cell myocarditis |
| Cytomegalovirus |
| Toxoplasmosis |
| Chagas |
| Rheumatic |
| Lyme |
| 2. Infiltrative |
| Amyloid |
| Sarcoid |
| Hemochromatosis |
| Carcinoid |
| Hypereosinophilic |
| Glycogen storage |
| Cardiac tumors |
| 3. Toxins |
| Doxorubicin |
| Chloroquine |
| Radiation injury |
| 4. Genetic |
| Fabry |
| Kearns-Sayre syndrome |
| Right ventricular dysplasia |
Several investigators have performed endomyocardial biopsies in patients with unexplained congestive heart failure (CHF) or ventricular arrhythmia.2,20,24 The percentage of patients with biopsies interpreted as myocarditis varied widely, primarily owing to the different diagnostic criteria for active myocarditis used by the investigators. This variability of endomyocardial biopsy criteria prompted a meeting of cardiac pathologists to reach a consensus on the pathologic definition of myocarditis, now known as the Dallas criteria.26 These criteria separate initial biopsies into myocarditis, borderline myocarditis, or no myocarditis. Active myocarditis is defined as “an inflammatory infiltrate of the myocardium with necrosis and/or degeneration of adjacent myocytes not typical of the ischemic damage associated with coronary artery disease” (Fig. 35–5). The term borderlinemyocarditis is applied when the inflammatory infiltrate is too sparse or myocyte injury is not demonstrated. Repeat biopsy is then suggested. A high frequency of active myocarditis is confirmed by repeat biopsy in patients whose initial histologic samples demonstrated borderline myocarditis. When RV endomyocardial biopsy has failed to establish the diagnosis, sampling the LV can improve diagnostic yield.
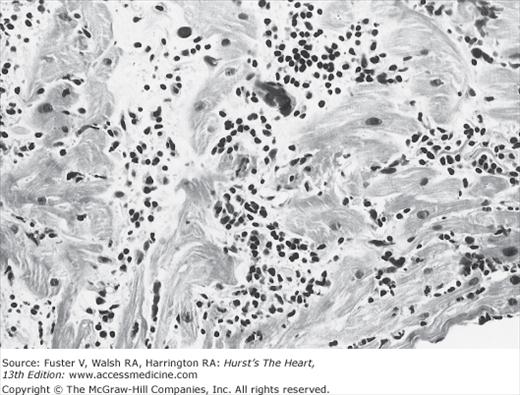
Although the Dallas criteria standardize the description of biopsy samples, histopathology alone can be inadequate to identify the presence of active myocarditis.27 Alternative classification schemes have been proposed, including one that combines histopathologic and clinical criteria.28 Myocarditis is divided into four subgroups—fulminant, acute, chronic active, and chronic persistent. These categories provide prognostic information and suggest which patients can or cannot benefit from immunosuppressive therapy (Table 35–3). Additionally, use of immunohistologic markers of inflammation, such as upregulation of histocompatibility leukocyte antigens (HLAs) on myocytes or detection of autoantibodies, can aid diagnosis as these changes are generalized and not focal.
| Fulminant | Acute | Chronic Active | Chronic Persistent | |
|---|---|---|---|---|
| Symptom onset | Distinct | Indistinct CHF, LV dysfunction | Indistinct CHF, LV dysfunction | Indistinct |
| Clinical presentation | Cardiogenic shock, severe LV dysfunction | Non-CHF symptoms, normal LV function | ||
| Initial biopsy | Multifoci of active myocarditis | Active or borderline myocarditis | Active or borderline myocarditis | Active or borderline myocarditis |
| Clinical natural history | Complete recovery or death | Incomplete recovery or dilated CM | Dilated CM | Non-CHF symptoms, normal LV function |
| Histologic natural history | Complete resolution of myocarditis | Complete resolution of myocarditis | Ongoing or resolving myocarditis, fibrosis | Ongoing or resolving myocarditis |
| Immunosuppressive therapy | No benefit | Sometimes beneficial | No benefit | No benefit |
Laboratory findings are generally not diagnostic. Leukocytosis, eosinophilia, and an elevated erythrocyte sedimentation rate (ESR) are sometimes present, as are elevated titers to cardiotropic viruses. A four-fold rise in immunoglobulin G (IgG) titer over a 4- to 6-week period is required to document acute infection. Elevated immunoglobulin M (IgM) antibody titer can denote an acute infection more specifically than a rise in IgG antibody titer. Unfortunately, a rise in antibody titer documents only the response to a recent viral infection and is not specific for active myocarditis. Abnormalities in peripheral T- and B-lymphocyte counts have been reported, but these findings have not been consistent and cannot be used as diagnostic adjuncts. An increase in the myocardial band (MB) of creatine phosphokinase (CPK) is observed in approximately 10% of patients, but troponin assays are proving to be more sensitive for detecting myocardial injury in patients with suspected myocarditis.29 The classic clinical triad of preceding viral illness, pericarditis, and associated laboratory abnormalities used to diagnose Coxsackie B–induced myocarditis is present in fewer than 10% of histologically proven cases.20
The ECG most frequently shows sinus tachycardia. Diffuse ST and T-wave changes, prolongation of the QTc, conduction delay, low voltage, and even an acute myocardial infarct pattern have been noted in patients with myocarditis. Cardiac arrhythmias are frequently observed, including complete heart block, ventricular tachycardia, and supraventricular arrhythmias—especially in the presence of CHF or pericardial inflammation.
Recent advances in cardiac MRI have demonstrated the potential for this technology to aid in the diagnosis of myocarditis and increase the sensitivity of endomyocardial biopsy. With one study, it is possible to assess LV function, tissue edema, inflammation, and scar, all of which may be present in patients with myocarditis. By identifying areas of myocardial inflammation, cardiac magnetic resonance imaging (MRI) facilitates selecting which patients should undergo endomyocardial biopsy and localizes areas of the myocardium that should be sampled. Mahrholdt and coworkers30 used contrast-enhanced MRI to identify areas of active inflammation in patients with clinically diagnosed myocarditis. Histopathologic analysis of endomyocardial biopsy samples taken from areas of enhancement revealed active myocarditis in 19 of 21 patients. Foci of inflammation were more frequently located on the lateral wall of the LV. Conversely, only one of 11 biopsy samples taken from regions lacking enhancement showed active myocarditis.
The International Consensus Group on Cardiovascular Magnetic Resonance in Myocarditis recently summarized the current state-of-the art use of cardiovascular magnetic resonance (CMR).31 With a caveat that their recommendations were based on a combination of expert opinion and small, single-center experiences, they put forth diagnostic CMR criteria for myocarditis, the Lake Louise Criteria. The authors recommended the combined use of three tissue markers: T2-weighted imaging looking for increased signal intensity, increased early gadolinium enhancement ratio between myocardial and skeletal muscle in T1-weighted images, and focal areas of subepicardial late gadolinium enhancement. The presence of at least two of these criteria in the setting of clinically suspected myocarditis can establish myocardial inflammation with a diagnostic accuracy of 78%. The authors further recommended a repeat study be considered for patients with an absence of any criteria and a recent onset of symptoms and for patients who fulfilled only one diagnostic criterion.
Echocardiography can reveal LV systolic dysfunction in patients with a normal-sized LV cavity. Segmental wall-motion abnormalities can be observed. Wall thickness can be increased, particularly early in the course of the disease, when inflammation is fulminant. Echocardiographic findings in active myocarditis can mimic restrictive, hypertrophic, or DCM. The presence of biventricular dysfunction is a strong predictor of death or transplantation.32
The cornerstone of therapy for patients with acute myocarditis is supportive care. Diuretics, angiotensin-converting enzyme (ACE) inhibitors, β-blockers, and aldosterone antagonists should be given in the proper clinical context. Intravenous inotropes should be reserved for patients with reduced cardiac output and signs of organ hypoperfusion. Digoxin can increase the expression of inflammatory cytokines and should be administered cautiously at a low dose.
When acute myocarditis presents with profound hemodynamic collapse, mechanical circulatory support devices can be used to bridge patients either to cardiac transplantation or to recovery. Although improvement in cardiac function has been reported to parallel the clearance of the inflammatory infiltrate, the duration of necessary device support has ranged from 7 to 70 days.33 Serial endomyocardial biopsy, echocardiography, right heart catheterization, and exercise testing with simultaneous hemodynamic and echocardiographic measurements are all used to determine native cardiac reserve and the suitability for device explantation.
Acceptance of the immune-mediated injury hypothesis has led many to question whether anti-inflammatory therapy additionally benefits patients with myocarditis treated with conventional heart failure regimens. Parillo et al34 studied 102 patients with DCM and classified them as being “reactive,” with endomyocardial or laboratory evidence of ongoing inflammation, or “nonreactive.” The study’s primary endpoint was an increase in LVEF equal to or greater than 5%. At 3 months of follow-up, 67% of reactive patients receiving prednisone reached this endpoint compared with only 28% in the reactive control group. After 9 months of follow-up, the prednisone-induced improvement in LVEF was no longer present. Nonreactive patients did not improve with prednisone. Despite these negative results, the authors concluded that prednisone therapy could provide modest improvements in clinical endpoints, but only in certain reactive subpopulations.
Anecdotal success with immunosuppression in active viral myocarditis led to the multicenter Myocarditis Treatment Trial.35 In this study, 111 patients with biopsy-proven myocarditis and LVEF less than 45% were randomized to receive conventional therapy alone versus immunosuppressive therapy with prednisone in combination with either azathioprine or cyclosporine. The study’s primary endpoint was a change in ejection fraction over 28 weeks. For all patients, the average increase in ejection fraction over baseline was 9%. Treatment did not improve LV ejection fraction, attenuation of clinical disease, or mortality (Fig. 35–6).
Figure 35–6
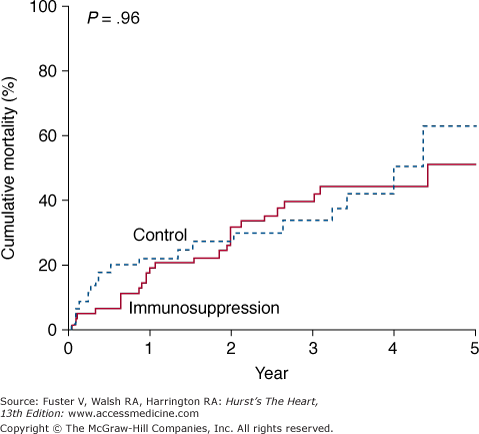
Actuarial mortality curves from the Myocarditis Treatment Trial illustrating no difference in survival between the treatment groups. Reproduced with permission from Mason et al.35
High-dose intravenous immune globulin (IVIG) has both immunomodulatory and antiviral effects. In a small, open-label study, nine of 10 adult patients with new-onset heart failure treated with IVIG had a significant improvement in LV function.36 When IVIG was tested in a prospective, placebo-controlled trial in 62 patients with recent-onset DCM and LVEF less than 40%, the results were disappointing.37 Although ejection fraction improved by 16% at 1 year in the IVIG-treated group, this increase was essentially equaled by those taking placebo. Thus, no benefit of immunomodulation could be demonstrated.
A more recent study examined the selected use of immunosuppressive therapy in patients with DCM and immunohistochemical evidence of inflammation.38 Eighty-four patients with DCM for at least 6 months who had increased HLA expression on endomyocardial biopsy specimens were randomized to receive standard heart-failure therapy alone or in combination with prednisone and azathioprine. After 2 years of follow-up, there was no difference in the composite primary endpoint of death, transplantation, or hospital readmission. The patients treated with immunosuppressive therapy, however, experienced a significant increase in ejection fraction and improvement in NYHA functional class at 3 and 24 months.
There is a growing understanding that differential responses to therapy are determined by whether there is ongoing immune activation or viral persistence in the myocardium. In one analysis, Frustaci and colleagues39 reported on a group of 41 patients with acute myocarditis treated with azathioprine and prednisone. Twenty-one patients were considered “responders” and experienced an average increase in LVEF from 25.7% to 47.4% at 1 year. The other 20, who were considered “nonresponders,” exhibited no change in LVEF (27.2%-26.5%). Viral genomes were present in 85% of the nonresponders but in only 14% of the responders. Conversely, cardiac autoantibodies were present in 91% of the responders and none of the nonresponders. Overall, the results suggest that immunosuppression may be helpful in patients with ongoing immune activation but is not likely to be effective in patients with viral persistence. This type of targeted immunosuppression was tested in the Tailored Immunosuppression in Inflammatory Cardiomyopathy (TIMIC) study, a randomized, controlled trial of immunosuppressive therapy in virus-negative myocarditis.40 The study’s investigators reported that compared with placebo, treatment with prednisone and azathioprine increased LVEF, improved NYHA functional class, and decreased LV dimensions in patients with active myocarditis and no viral persistence. Removal of circulating cardiac autoantibodies through the use of immunoadsorption may also be effective, as demonstrated in a small, single-center trial.41
For patients with ongoing viral genomic expression, treatment with interferon may be of some benefit. Interferon-α (INF-α) has been described as producing hemodynamic and clinical improvement for patients with viral myocaridits.42 Another small trial investigated whether administration of interferon-β (INF-β) to patients with LV dysfunction and persistence of viral genome expression could safely eliminate viral presence and improve myocardial function.43 Twenty-two patients with persistent adenoviral or enteroviral genomes from endomyocardial biopsies and worsening heart failure symptoms refractory to standard medical therapy were treated for 6 months with INF-β. Treatment produced structural and functional LV improvement, and a majority of patients enjoyed a reduction in NYHA functional class. Viral genome was successfully eliminated in all patients, and myocardial inflammation was significantly decreased. A large prospective trial is underway to confirm these preliminary results.
Taken as a whole, these trials do not support the routine use of immunosuppressive therapy in patients with myocarditis. However, present data suggest that subgroups with ongoing myocarditis and cardiac autoantibodies are more likely to benefit from immunosuppression, although no uniform methodology yet exists to identify them.
Approximately one-third of those who present with clinical carditis and recover will be left with some cardiac abnormality, ranging from mild changes on ECG to significant heart failure. Approximately 40% of all patients will completely recover.20 Currently, no clinical criteria can reliably predict who will recover, although the vast majority of patients with mild reductions in LVEF and NYHA class I or II heart failure recover completely. In one study, a pulmonary capillary wedge pressure (PCWP) greater than 15 mm Hg was a univariate predictor of death or need for cardiac transplantation.44 In multivariate analysis, PCWP and histopathology of lymphocytic, granulomatous, or giant cell myocarditis each significantly predicted mortality or need for transplant. Paradoxically, patients with fulminant myocarditis have an excellent long-term prognosis despite their experience of circulatory collapse. In one study, long-term transplant-free survival was 93% compared with only 45% for those with acute myocarditis.21 Elevated levels of IL-10 can identify a subgroup of patients with fulminant myocarditis who are at higher risk of developing cardiogenic shock with the need for mechanical circulatory support and those at higher risk of mortality.45
The prognosis of patients with myocarditis depends to some extent on the causative agent, but for patients with histopathologic confirmation of myocarditis, the 1-year survival rate is approximately 80%, and 5-year survival is in the range of 50% to 60%.44 Chronic inflammation, viral persistence, or both can affect disease progression and prognosis. Future therapies will need to identify the predominant factor to target treatment and hopefully improve survival.
American trypanosomiasis, or Chagas disease, is the most common cause of CHF in rural South and Central America. This condition results from the bite of the reduviid bug, leading to infection with Trypanosoma cruzi. The pathogenesis of chronic chagasic cardiomyopathy is controversial because the parasite is rarely present in the myocardium. As in the viral cardiomyopathy model, the cardiac injury is thought to be immunologically mediated.46 Both cellular and humoral immune responses have been implicated in the myocardial injury.
This parasitic disease has an acute phase during which hematogenous spread of the parasite leads to invasion of various tissues and organ systems. The invasion is accompanied by an intense inflammatory reaction with mononuclear cells and is characterized by fever, sweating, myalgias, myocarditis, hepatosplenomegaly, and a case fatality rate of approximately 5%. Survivors enter an asymptomatic latent phase, but 20% to 30% of them develop a chronic form of the disease up to 20 years after the initial infection.
The chronic stage is a result of gradual tissue destruction. The gastrointestinal tract and heart are the most common sites of involvement, with the primary cause of death being cardiac failure. In the gut, the destruction of the myenteric plexus is responsible for the development of megaesophagus and megacolon. In the heart, the myofibrils and the Purkinje fibers are replaced by fibrous tissue, leading to cardiomegaly, CHF, heart block, and arrhythmia.
Diagnosis of the acute disease depends on the discovery of trypomastigotes in the blood of the infected individual. In chronic infection, direct diagnosis is less useful because there are fewer circulating trypomastigotes. Xenodiagnosis (in which the patient is bitten by reduviid bugs bred in the laboratory and the parasite is subsequently identified in the intestine of the insect) is the most useful test, which detects infection in approximately 50% of patients. The complement-fixation test (Machado-Guerreiro test) also has high sensitivity and specificity for identification of chronic Chagas disease. In the other laboratory tests, it is necessary to rely on positive serologic test results (eg, the indirect immunofluorescent antibody, enzyme-linked immunosorbent assay, and hemagglutination tests) together with symptoms and signs compatible with Chagas disease.
Endomyocardial biopsy can show active myocarditis using the Dallas criteria. Acute echocardiography may identify pericardial effusions during acute Chagas myocarditis. In chronic Chagas cardiomyopathy, segmental wall motion abnormalities may be identified, and more than half of symptomatic patients will have LV apical aneurysms.47 ECG findings include complete heart block, AV block, or right bundle-branch block with or without fascicular block. Myocardial delayed enhancement by cardiac MRI is an accurate method for identifying the myocardial fibrosis seen in Chagas cardiomyopathy.48
The treatment of patients with chronic Chagas disease is symptomatic and includes a pacemaker for complete heart block, an implantable cardioverter-defibrillator for recurrent ventricular arrhythmia, and standard therapy for CHF as outlined for other forms of myocarditis. Antiparasitic agents such as nifurtimox and benznidazole eradicate parasitemia during the acute phase and are typically curative. They should be administered if the disease has not previously been treated and can be used as prophylaxis if there is a high likelihood of recurrence, such as after immunosuppressive therapy. A large prospective trial is currently underway to help determine whether trypanocidal therapy with benznidazole is effective to alter clinical outcomes for patients with chronic Chagas cardiomyopathy. The role of immunosuppression therapy for chagasic myocarditis is controversial, and heart transplantation is effective for end-stage refractory cardiac disease. Cases of reactivation may occur, but specific therapy can reverse ventricular dysfunction.49
A prognostic scoring system based on six clinical risk factors has recently been published.50 These include NYHA class III or IV, cardiomegaly by radiography, LV wall motion abnormalities, nonsustained ventricular tachycardia detected by Holter monitoring, low QRS voltage, and male gender. Ten-year mortality was 9%, 37%, and 85% for those at low, intermediate, and high risk, respectively. The use of this prognostic scoring system can aid in directing resources to patients at highest risk, especially in regions of the world where access to care can be limited.
Lyme disease results from infection with the spirochete Borrelia burgdorferi, which is introduced by a tick bite. The initial presenting symptom in patients with the disease who progress to cardiac involvement is frequently complete heart block. Endomyocardial biopsy may show active myocarditis; spirochetes are rarely seen on biopsy. Corticosteroid administration is helpful in treating patients with Lyme carditis after therapy with tetracycline.
Among other infectious causes is Toxoplasma gondii, which is curable by pyrimethamine and sulfadiazine and occurs most commonly in immune-deficient hosts. Leptospirosis is another common cause in fatal cases of myocarditis. Fifty percent of patients have ST- and T-wave changes on ECG.
Acute rheumatic fever can occur in children and young adults. It generally occurs after a group A streptococcal pharyngitis, but only indirect evidence linking the two has been found. Rheumatic carditis may result from a direct toxic effect of some streptococcal product versus an immunologic mechanism.51 Group A streptococci have a number of structural components similar to those of human tissue. Antibodies to streptococci cross-react with the glycoproteins of heart valves. The serum of patients with rheumatic fever contains autoantibodies to myosin and sarcolemma. The Aschoff body, which is pathognomic for this disorder, represents persistent focal inflammatory lesions in the myocardium. These can persist for years after an acute attack. Macrophages containing myosin have been identified in these nodules.
Clinical diagnosis is made using the Jones criteria52 The major manifestations are carditis, polyarthritis, chorea, erythema marginatum, subcutaneous nodules, and evidence of preceding streptococcal infection (ie, positive throat culture, history of scarlet fever, elevated antistreptolysin titers). Minor criteria are nonspecific findings such as fever, arthralgia, previous rheumatic fever or rheumatic heart disease, elevated ESR or C-reactive protein, and prolonged PR interval. Diagnosis is made by the presence of two major criteria or one major and two minor criteria.
Two-thirds of patients present with an antecedent pharyngitis followed by the symptoms of rheumatic fever in 1 to 5 weeks, with a mean presentation of 18.6 days. Severe carditis resulting in death can occur but is unusual. CHF is observed in only 5% to 10% of patients. The carditis is usually mild, with scarring of the heart valves more typically predominant. Physical examination is notable for fever and heart murmurs, reflecting the acute valvulitis. The mitral valve is involved three times as frequently as the aortic valve; therefore, mitral murmurs are more common. Mitral regurgitation is the most common finding. A mid-diastolic murmur over the apical area can frequently be heard. This is called the Carey Coombs murmur, and its presence almost certainly confirms mitral valvulitis. Aortic insufficiency can be auscultated with aortic valvulitis.
There are no characteristic ECG findings, although PR prolongation and nonspecific ST-T-wave changes are frequently described. Endomyocardial biopsy demonstrates the Aschoff body as well as a diffuse cellular interstitial infiltrate including lymphocytes, polymorphonuclear cells, histiocytes, and eosinophils. Laboratory tests suggestive of rheumatic fever include antibodies to antistreptolysin O and anti-DNAse B, an elevated ESR, and elevated C-reactive protein. Extracardiac manifestations generally include an acute migratory polyarthritis of the large joints.
Aspirin and penicillin are the mainstays of therapy. Corticosteroids can also provide symptomatic relief. Treatment with IVIG produces no detectable clinical or echocardiographic improvements.53 Mitral valve repair during acute carditis is associated with an increased mortality risk and should be undertaken only when heart failure is refractory to optimal anti-inflammatory therapy.
After rheumatic fever has been diagnosed, antibiotic prophylaxis is required to prevent recurrent episodes. The most effective method is a single monthly intramuscular injection of 1.2 million units of benzathine penicillin G until age 21 years.



