Mycotic and Actinomycetic Infections of the Lung
Shari L. Meyerson
David A. Harpole Jr.
Fungal infections of the lungs have traditionally represented a very small component of the practice of most thoracic surgeons. Most fungal pathogens are opportunistic, causing clinically significant infection only in the presence of impaired host defenses. The exception is a trio of endemic fungi which commonly cause disease in normal hosts. These are a significant clinical issue for surgeons in the Ohio and Mississippi River Valleys, where histoplasmosis is endemic in the Midwest and blastomycosis is endemic in the South and Southeast, and the Sonoran desert regions of the southwest United States, where coccidioidomycosis is endemic. In the immunocompetent patient, the surgeon’s involvement in fungal infections generally consists of diagnosis or, more importantly, differentiation from cancer, which fungal nodules will often resemble. Unfortunately, the incidence of severe invasive fungal infections is growing as the population of immunologically compromised patients increases. This group includes patients at the extremes of age, diabetics, patients receiving corticosteroids for a variety of illnesses, organ transplant recipients, cancer patients undergoing chemotherapy, and patients with acquired immune deficiency syndrome. Immunocompromised patients can not only develop more severe disease related to the above three fungi but are also susceptible to a large number of fungi that rarely cause disease in the normal host.
Pulmonary involvement may be the primary site of infection or may be simply a component of a disseminated systemic infection. Fungal organisms are notoriously difficult to grow in culture without adequate tissue samples. Therefore the thoracic surgeon can play a role in the diagnosis of a systemic fungal infection with multiple pulmonary nodules. Conversely, fungal infections can also present as a single solid or cavitary lesion for which surgery will have a therapeutic role as well. The typical presentation, distribution of involvement, and recommended treatment are strongly dependent on the specific type of fungus. The most common fungal infections encountered by the practicing thoracic surgeon are discussed here, with special emphasis on the spectrum of pulmonary disease and the role of operative intervention.
Antifungal Medications
Antifungal therapy is a rapidly changing field with the continued development of new drugs within existing classes as well as novel classes of antifungal agents. Individual antifungal agents and their doses in specific fungal infections are discussed with the appropriate fungal organisms. However, it is appropriate to briefly summarize the major categories of antifungal agents currently in clinical use. The major classes, available drugs, mechanisms of action, and available formulations are presented in Table 92-1 and discussed below.
Amphotericin B is a polyene drug—the first drug developed for the treatment of invasive fungal infections. It is a product of the actinomycete Streptomyces nodosa. Polyenes act by binding to ergosterol, a component of fungal cell membranes, causing cell disruption. Amphotericin B is poorly absorbed by the gastrointestinal tract and is effective only via the intravenous route. Amphotericin B is associated with significant toxicities, which limit its usage. During infusion, fevers, rigors, and hypotension are not uncommon. Significant nephrotoxicity is also noted, which is the most common limiting factor for amphotericin B total dose. Amphotericin B is also available in lipid formulations, which, although significantly more expensive, are much better tolerated, with decreased nephrotoxicity.
First-generation triazoles, including fluconazole and itraconazole, were developed in the 1990s. Triazoles act by inhibiting the cytochrome P-450 enzyme, which converts lano- sterol to ergosterol in the fungal cell membrane. First-generation triazoles are available in intravenous and oral formulations and are generally well tolerated. They still remain first-line therapy for less severe infections of various types. Unfortunately widespread usage has led to the development of resistance. The second-generation triazoles—including voriconazole, posaconazole, and the investigational drug ravuconazole—appear to have significant activity against a wide range of fungi and are gaining an increasing role as first- and second-line therapy.(Espinel-Ingroff 2001)11,13 Their primary toxicity is related to mild hepatic dysfunction and interactions with other drugs metabolized by the hepatic cytochrome P-450 enzyme, such as warfarin, cyclosporine, phenytoin, and rifampicin.
Echinocandins are amphiphilic lipopeptides formed during the fermentation of some fungi. They act by a completely different mechanism from the other classes of antifungals. Echinocandins inhibit (1,3)-β-D-glucan synthase, which produces an important component of the fungal cell wall.52 This target exists in many species of Aspergillus and Candida but not in the cell wall
of Cryptococcus species. Importantly, this target does not exist in human cells; therefore the toxicity of the echinocandins is minimal, with the most prominent side effects including headache, fever, and reversible hepatic enzyme elevation.
of Cryptococcus species. Importantly, this target does not exist in human cells; therefore the toxicity of the echinocandins is minimal, with the most prominent side effects including headache, fever, and reversible hepatic enzyme elevation.
Table 92-1 Currently Available Antifungal Medications | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
Infections in the Normal Host Commonly Caused by Fungi
Histoplasmosis
Histoplasma capsulatum, an airborne spore, is responsible for more pulmonary infections than any other fungus. It exists throughout the world but is most commonly seen in North America and Central America. In the United States, the organism is endemic in the Ohio and Mississippi River valleys (Fig. 92-1). It is found in the soil and grows best in areas with large amounts of bird or bat guano. In fact, the fungus can thrive for years in contaminated soil even if the birds or bats are removed from the area. Disseminated infection was first described by Samuel Darling just over a century ago while he was working in Panama. He described a fatal case of disseminated disease from Martinique.15 Histoplasmosis in the early half of the 1900s was thought to be a rare and almost always fatal disease. It was not until the 1940s, with the development of the histoplasmin skin test antigen, that the relationship between histoplasmosis and pulmonary calcifications was established. (Palmer 1945) Large-scale studies by the U.S. Public Health Service demonstrated positive skin test reactivity in over 80% of young adults from the areas surrounding the Mississippi and Ohio Rivers. The vast majority had no clinical symptoms and no idea that they had ever been exposed to or infected with a fungus. Overall it is estimated that <1% of patients infected with H. capsulatum will develop symptoms. Most often histoplasmosis is diagnosed much later, with the finding of calcified lung lesions or mediastinal lymph nodes indicative of previous infection. These late findings can raise concern for malignancy and many of these patients will come to the attention of the thoracic surgeon. A solitary pulmonary nodule due to histoplasmosis has a classic appearance. There is a small necrotic center initially and, over many years, concentric layers of collagen form around this stimulus. These will generally exhibit central or concentric laminar calcification. Lesions due to old histoplasmosis will not be hypermetabolic on positron emission tomography (PET) scanning. Identification of this classic radiographic picture is generally sufficient to determine the benign nature of the lesion. Lesions that are not calcified should be further investigated with biopsy or surgical resection to rule out malignancy.
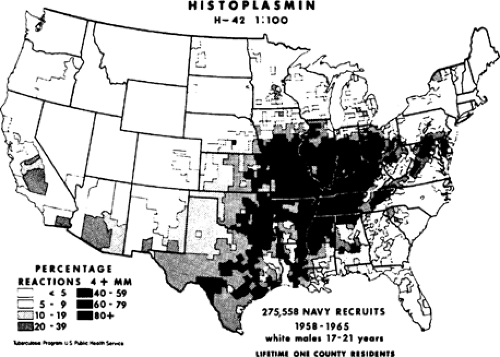 Figure 92-1. Map of the United States showing areas of endemicity for histoplasmosis. (From Edwards LB et al. An atlas of sensitivity to tuberculin, PPD-B and histoplasmin in the United States. Am Rev Respir Dis 1969;99(Suppl):1. With permission.) |
The clinical manifestations of histoplasmosis can be divided into several syndromes: acute pulmonary histoplasmosis, chronic cavitary pulmonary histoplasmosis, mediastinal granuloma, mediastinal fibrosis, and disseminated histoplasmosis. Each syndrome has a different clinical picture, time course, and treatment.
Acute pulmonary histoplasmosis is usually a self-limited illness occurring at the initial exposure to the organism. Symptoms are similar to those of other community-acquired pneumonias, with fever, dry cough, weakness, malaise, chest discomfort, and mild dyspnea. Chest x-ray shows patchy pneumonia involving one or more lobes, often with enlarged hilar and mediastinal lymph nodes (Fig. 92-2). In immunocompetent patients, the symptoms are self-limiting and resolve fairly rapidly. The fatigue, however, can linger for several months. Some 5% of patients
will have joint and/or skin manifestations. Skin manifestations include erythema nodosum and erythema multiforme, which are most commonly seen in young women. Above and beyond the commonly reported joint and muscle pains of the acute infection, a small minority of patients will develop a polyarticular symmetrical arthritis. The joint symptoms can last several weeks beyond the pulmonary symptoms.
will have joint and/or skin manifestations. Skin manifestations include erythema nodosum and erythema multiforme, which are most commonly seen in young women. Above and beyond the commonly reported joint and muscle pains of the acute infection, a small minority of patients will develop a polyarticular symmetrical arthritis. The joint symptoms can last several weeks beyond the pulmonary symptoms.
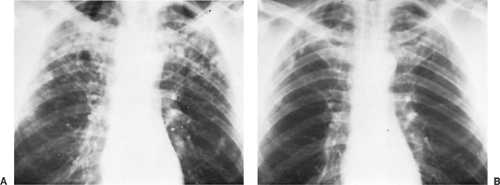 Figure 92-2. Acute histoplasmosis. A: Chest x-ray of an asymptomatic 48-year-old man with bilateral infiltrates. Sputum cultures were positive for Histoplasma capsulatum. B: Same patient 2 years later, without intervening treatment, showing only minimal residual nodules. |
Two situations can change the picture of acute pulmonary histoplasmosis. If the inoculum is extremely large, as when large amounts of dust from a contaminated bat cave are inhaled, the clinical picture becomes severe. The onset of symptoms is very rapid, with fever, chills, cough, and chest pain. Diffuse nodular infiltrates are seen throughout both lungs on radiographs. This can progress to adult respiratory distress syndrome within days. A similar clinical picture will be seen in the immunocompromised patient, although the onset and progression are not as rapid. Patients with severe disease should be treated with antifungal agents to limit respiratory compromise and speed recovery.
As the acute disease resolves, scattered nodules will be left throughout the involved areas. Over time, these will often calcify, producing the characteristic lesions of healed histoplasmosis. Similar calcifications will appear in involved mediastinal lymph nodes (Fig. 92-3). Over many years, these calcified lymph nodes can erode into an adjacent bronchus, causing inflammation, obstruction, and occasionally broncholithiasis, where the patient will develop a cough productive of small amounts of calcified, gravel-like material. Bronchoscopic retrieval of obstructing broncholiths can be required to resolve postobstructive pneumonias.
Patients can also go on to develop chronic cavitary histoplasmosis. This is an epidemiologically interesting phenomenon because the chronic cavitary form is nearly always associated with older patients in the setting of underlying pulmonary disease, specifically emphysema.29 Cavities develop when the fungal infection is near emphysematous bullae, usually in the apices of the lungs. The walls of the bullae thicken and then develop necrosis, leading to cavitary lesions. Necrosis can continue to progress, leading to enlarging cavities as well as other complications, including bronchopleural fistula and pneumothorax. Clinically these patients report productive cough, dyspnea, mild hemoptysis, fatigue, and anorexia, all of which are similar to symptoms of exacerbation of their underlying emphysema. The differential diagnosis includes tuberculosis and atypical mycobacterial infections as well as cavitary lung cancer, as most of these patients have an extensive smoking history (Fig. 92-4). If untreated, chronic cavitary histoplasmosis can lead to death due to progressive respiratory insufficiency in an already damaged lung.25
Histoplasmosis is associated with significant mediastinal lymph node involvement. In most patients, as the infection resolves, the enlarged mediastinal lymph nodes will eventually
shrink and calcify. Among patients with histoplasmosis, <10% will develop massive inflammation of mediastinal nodes with caseating necrosis. Some are asymptomatic, but others can develop airway obstruction, vascular obstruction, or esophageal obstruction due to the mass effect of these nodes. Symptoms can occur up to years after the primary infection. Occasionally fistulas can develop between the nodes and mediastinal structures. These nodes will usually regress without treatment, but the mass effects and symptoms can persist for years.59 Occasionally surgical intervention is required for debulking of the nodes when obstructive symptoms become severe.19
shrink and calcify. Among patients with histoplasmosis, <10% will develop massive inflammation of mediastinal nodes with caseating necrosis. Some are asymptomatic, but others can develop airway obstruction, vascular obstruction, or esophageal obstruction due to the mass effect of these nodes. Symptoms can occur up to years after the primary infection. Occasionally fistulas can develop between the nodes and mediastinal structures. These nodes will usually regress without treatment, but the mass effects and symptoms can persist for years.59 Occasionally surgical intervention is required for debulking of the nodes when obstructive symptoms become severe.19
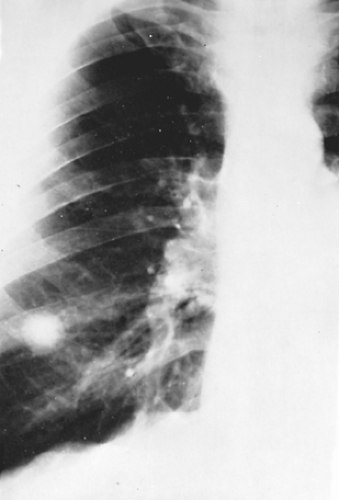 Figure 92-3. Residual calcified mediastinal adenopathy in an adult with previous histoplasmosis. |
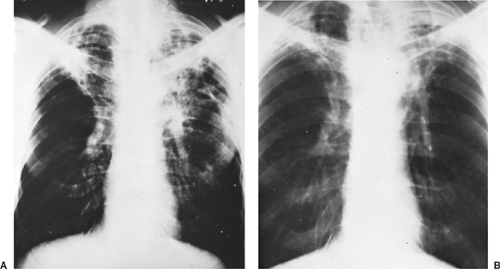 Figure 92-4. Chronic cavitary histoplasmosis. A: Chest x-ray showing cavitary lesions in the setting of sputum cultures positive for Histoplasma capsulatum. B: Chest x-ray of the same patient after 3 months of treatment with amphotericin B. |
Mediastinal involvement can also be seen in the form of fibrosing mediastinitis. Although this was originally thought to represent the end stage of mediastinal granuloma, current research suggests that these are two separate processes.16 Mediastinal fibrosis is thought to be caused by an excessive fibrotic response to H. capsulatum antigens rather than an ongoing infection. Organisms can rarely be isolated from the fibrotic tissue and there is no proven benefit to antifungal therapy. Fibrosing mediastinitis is more common in younger patients, with the peak incidence between age 20 and 40. A dense, fibrotic response envelops not only the mediastinal nodes but also any surrounding structures with mature collagen rather than the granulomas characteristic of acute infection. The fibrotic tissue can obstruct major mediastinal vascular structures, including the main pulmonary arteries, pulmonary veins, superior vena cava or even the right atrium. Other commonly involved structures include the right and/or left main bronchi and esophagus. Fibrosis is slowly progressive over years and does not regress. It is not responsive to either antifungal medications or cortico- steroids. Endobronchial and endovascular interventions, including dilation and stenting, can be helpful in improving symptoms at least temporarily. If mediastinal structures are involved bilaterally, the disease is almost uniformly fatal. If involvement is unilateral, pneumonectomy can be considered. While it can be lifesaving, in this setting pneumonectomy carries a high operative mortality due to dense adhesions and collateral blood supply.
Disseminated histoplasmosis is the most severe manifestation of infection. Most patients with histoplasmosis will have asymptomatic hematogenous dissemination. In patients with a normal immune system, H. capsulatum antigens are recognized and macrophages activated to kill the organism. The organism is generally not completely eradicated and will persist in small foci in various organs for many years. As with tuberculosis, this can lead to reactivation years later, especially if immunosuppression develops. In the immunosuppressed patient, inadequate cell-mediated immunity allows the development of symptomatic disease at the time of acute infection or via reactivation of latent disease. Risk factors include young age, AIDS, transplant, hematologic malignancies, and congenital T-cell deficiencies. Disseminated disease can manifest with vague systemic symptoms, skin and mucosal lesions, central nervous system involvement, and endocarditis. One site of dissemination seen in histoplasmosis more commonly than in other fungal diseases is the adrenal gland. Addison’s disease can be seen, with vascular occlusion and necrosis of bilateral adrenal glands.
Diagnosis of histoplasmosis is generally made by culture of tissue or body fluids. Tissue biopsy in the acutely ill patient can demonstrate the characteristic 2- to 4-μm oval, narrow-based budding yeasts (Fig. 92-5). These look similar to Candida and Leishmania but can generally be differentiated on clinical grounds. Urine antigen tests can also detect the organism but are only positive with a high disease burden. Skin tests exist for Histoplasma as well, but there is extensive cross reactivity with other fungal organisms.
The treatment of histoplasmosis depends on the clinical syndrome (Table 92-2). Acute histoplasmosis does not generally
require treatment. Most patients will recover within 4 to 6 weeks. Patients who are still symptomatic after 4 to 6 weeks can be treated with oral itraconazole (200 mg once or twice daily) for 6 to 12 weeks.69 Patients with severe acute pulmonary histoplasmosis with hypoxemia should be treated with amphotericin B until clinical improvement begins and then switched to itraconazole (200 mg twice daily) for 3 to 6 months. Chronic cavitary histoplasmosis should be treated with itraconazole (200 mg twice daily) to prevent progression to respiratory failure. Total length of treatment may need to be 12 to 24 months, with the potential for relapse after that. Mediastinal granuloma is treated with oral itraconazole (200 mg once or twice daily) for 6 to 12 months based on the premise that there is ongoing granulomatous inflammation, which should respond to antifungal therapy. There is little evidence to support this outside of anecdotal reports. Severe obstructive symptoms are treated with amphotericin B until symptoms improve, followed by itraconazole for 6 to 12 months.69 If obstructive symptoms do not respond to antifungal therapy, surgical debulking may be attempted. Mediastinal fibrosis does not respond to medical therapy. The mainstay of treatment is endobronchial and endovascular dilation and stenting to decrease symptoms. Surgical debulking of the fibrotic tissue to relieve tracheal or bronchial compression can be performed but is associated with high operative mortality and complication rates.44 Disseminated histoplasmosis should be treated initially with amphotericin B. The lipid formulations available are associated with less toxicity, more rapid symptom resolution, and improved survival.34 Once symptoms improve, generally after a few weeks of therapy, treatment can be converted to oral itraconazole for 6 to 18 months.
require treatment. Most patients will recover within 4 to 6 weeks. Patients who are still symptomatic after 4 to 6 weeks can be treated with oral itraconazole (200 mg once or twice daily) for 6 to 12 weeks.69 Patients with severe acute pulmonary histoplasmosis with hypoxemia should be treated with amphotericin B until clinical improvement begins and then switched to itraconazole (200 mg twice daily) for 3 to 6 months. Chronic cavitary histoplasmosis should be treated with itraconazole (200 mg twice daily) to prevent progression to respiratory failure. Total length of treatment may need to be 12 to 24 months, with the potential for relapse after that. Mediastinal granuloma is treated with oral itraconazole (200 mg once or twice daily) for 6 to 12 months based on the premise that there is ongoing granulomatous inflammation, which should respond to antifungal therapy. There is little evidence to support this outside of anecdotal reports. Severe obstructive symptoms are treated with amphotericin B until symptoms improve, followed by itraconazole for 6 to 12 months.69 If obstructive symptoms do not respond to antifungal therapy, surgical debulking may be attempted. Mediastinal fibrosis does not respond to medical therapy. The mainstay of treatment is endobronchial and endovascular dilation and stenting to decrease symptoms. Surgical debulking of the fibrotic tissue to relieve tracheal or bronchial compression can be performed but is associated with high operative mortality and complication rates.44 Disseminated histoplasmosis should be treated initially with amphotericin B. The lipid formulations available are associated with less toxicity, more rapid symptom resolution, and improved survival.34 Once symptoms improve, generally after a few weeks of therapy, treatment can be converted to oral itraconazole for 6 to 18 months.
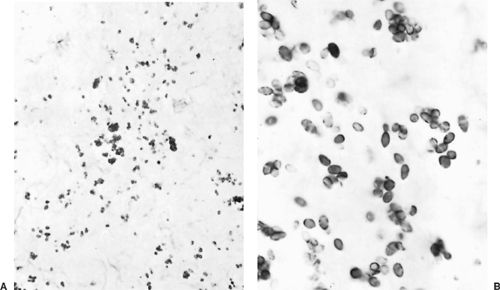 Figure 92-5. Histoplasma capsulatum organisms. A: Nonviable capsules in a necrotic area of a granuloma (Gomori’s stain, original magnification ×780). B: Viable yeast forms in a lymph node (original magnification ×1300). |
Table 92-2 Recommended Therapy for Histoplasmosis Based on Clinical Syndrome | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||
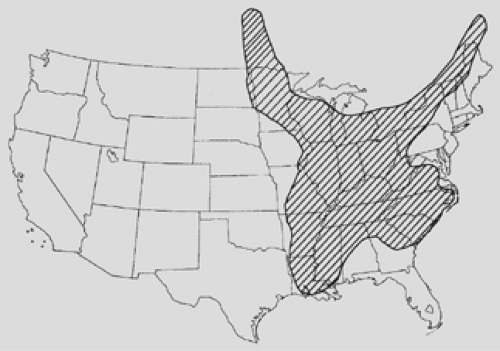 Figure 92-6. Map of the United States showing areas of endemicity for blastomycosis. (Adapted from Menges RW et al. Clinical and epidemiological studies on 79 canine blastomycosis cases in Arkansas. Am J Epidemiol 1965;81:169.) |
Blastomycosis
Blastomycosis is caused by the thick-walled fungus Blastomyces dermatitidis. This fungus is found in soil and is most common in the southeastern and south central states bordering the Mississippi and Ohio River valleys. It is also found in a small area of New York and Canada along the St. Lawrence River (Fig. 92-6). Blastomycosis was originally described in 1894 by Gilchrist.28 He described a skin infection caused by a protozoan organism. He did further work on this subject and eventually isolated the organism responsible and named the fungus B. dermatitidis. Although the cutaneous manifestations can be impressive, as Gilchrist originally described, it is now understood that the primary site of infection is the lungs, via inhalation of the organism, and skin involvement occurs with systemic dissemination.
Inhalation of B. dermatitidis triggers mechanisms of natural resistance mediated by neutrophils, monocytes, and alveolar macrophages.20 Macrophages can also inhibit conversion of the fungus from the mycelial form found in the environment to the yeast form found in tissue. This may be a significant part of the host defense, as the yeast form is more resistant to phagocytosis.65 Blastomyces is not generally an opportunistic pathogen and most infections occur in patients without immune deficiency. The typical patient is a young to middle-aged male with a history of outdoor work or recreation in one of the endemic areas.
The most common presentation is pulmonary involvement. Patients will often have nonspecific systemic symptoms such as weight loss, fever, fatigue, and cough. Radiologic findings most often include either an alveolar infiltrate or a mass-like infiltrate. Because blastomycosis is uncommon, these pulmonary findings are usually diagnosed as either a community-acquired pneumonia or a lung cancer initially. One common route to diagnosis of blastomycosis is growth of the fungus from bronchial washings or brushings taken at the time of bronchoscopy for the evaluation of a presumed lung cancer or persistent pneumonia unresponsive to antibiotics. Blastomycosis can also present a picture of miliary disease or reticulonodular infiltrates. Although cavitary lesions are sometimes seen, they are not as common as with histoplasmosis or tuberculosis (Fig. 92-7).
The next most common clinical presentation is cutaneous involvement. Cutaneous involvement can be seen with or without simultaneous radiologic evidence of pulmonary involvement. Skin lesions have a raised irregular border with crusting and some purulent drainage (Fig. 92-8). These lesions are overlying a subcutaneous abscess. If the abscess drains spontaneously, the lesion will become ulcerative with heaped-up edges and exudative material at the base. Aspiration of a subcutaneous abscess or biopsy of an ulcer is the diagnostic test most likely to yield the organism. Although pulmonary and cutaneous manifestations are most common, nearly any organ system can be involved. Other common sites include bone with osteomyelitis, genitourinary with prostatitis, central nervous system with meningitis, or epidural abscesses and head and neck with laryngeal ulcers.
Diagnosis of blastomycosis is usually made by identification of the fungus. Microscopic examination of fluid or tissue specimens demonstrates the characteristic 8- to 15-μm yeast cells with a thick cell wall (Fig. 92-9). Reproduction is by a single broad-based bud that grows to the size of the original organism prior to separating. B. dermatitidis is not particularly difficult to culture by fungal standards and will generally grow in 2 to 4 weeks. Unlike many other fungi, such as Aspergillus or Candida, colonization with B. dermatitidis does not occur. Identification of the organism in either tissue sections or culture reliably defines a pathologic infection. Serologic and skin tests are less helpful. Serologic techniques are limited by extensive cross reactivity with other fungi and lack of clinical availability of more sensitive techniques, such as enzyme immunoabsorbent assays. Skin tests have been used in the past but are commonly negative even in the setting of culture-proven infection. Blastomycosis skin tests are no longer available for clinical use.
Blastomycosis should be treated with antifungal medications whenever it is diagnosed. The mortality rate of untreated blastomycosis can be over 50%. For patients with mild to moderate disease, the recommended therapy is itraconazole 200 mg once or twice daily for 6 to 12 months. Fluconazole and ketoconazole appear to have less activity clinically. In severe or life-threatening disease, amphotericin B 0.7, 1 mg/kg per day, should be used until the patient improves clinically. Therapy can then be switched to oral itraconazole for 6 to 12 months.36 If the diagnosis is made at the time of thoracotomy for suspected malignancy, a full course of antifungal therapy should be given postoperatively. Surgical resection is occasionally indicated for known cavitary lesions of blastomycosis that do not respond to adequate trials of antifungal therapy. Even if sputum cultures are negative, fungal organisms can often be recovered from tissue culture of the cavity wall.
Coccidioidomycosis
Coccidioidomycosis is caused by the dimorphic fungus Coccidioides immitis. This fungus is endemic to parts of North and Central America, specifically the Sonoran Desert region (Fig. 92-10). The areas of highest concentration are the southern San Joaquin Valley of California and southern Arizona. C. immitis grows in the soil of endemic areas, producing small conidia that easily become airborne. The portal of entry is the respiratory system, and epidemics have been reported after severe dust
storms. Once in the lower respiratory tract, the conidia turn into multinucleated structures called spherules. These can often be identified in tissue sections of infectious nodules (Fig. 92-11). The spherule then produces up to hundreds of endospores. Once the endospores are mature, they are released into the tissue and the life cycle begins again. Most cases of coccidioidomycosis are seen in otherwise healthy patients; however, severe infections can be seen in immunocompromised patients. Certain racial groups have been identified as being at higher risk for more severe or disseminated disease, including individuals of Filipino ancestry and African Americans.12 The cause for this increased risk has not been identified.
storms. Once in the lower respiratory tract, the conidia turn into multinucleated structures called spherules. These can often be identified in tissue sections of infectious nodules (Fig. 92-11). The spherule then produces up to hundreds of endospores. Once the endospores are mature, they are released into the tissue and the life cycle begins again. Most cases of coccidioidomycosis are seen in otherwise healthy patients; however, severe infections can be seen in immunocompromised patients. Certain racial groups have been identified as being at higher risk for more severe or disseminated disease, including individuals of Filipino ancestry and African Americans.12 The cause for this increased risk has not been identified.
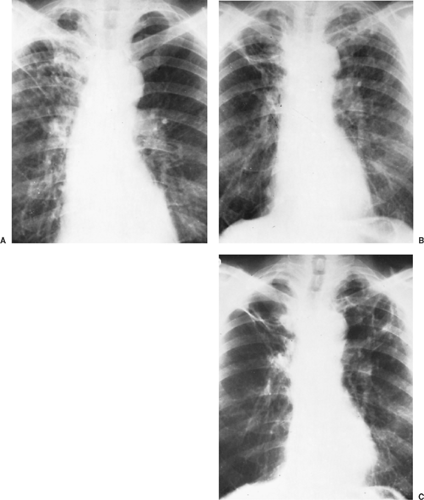 Figure 92-7. Evolution of chronic pulmonary blastomycosis. A: Chest x-ray showing extensive right upper lung infiltrative lesion in a patient with proven cutaneous blastomycosis. B: Chest x-ray 4 years later, without treatment, showing progression to bilateral cavitary disease. C: Chest x-ray 2 years later, after treatment, showing residual thin-walled cavities. |
The majority (60%) of patients infected are asymptomatic. Several clinical syndromes, however, are associated with acute C. immitis infection. The best known is valley fever, a mild to moderate flu-like illness, which is seen in up to 40% of acute infections. Prominent symptoms include fevers, cough, weight loss, night sweats, and pleuritic chest pain. When arthralgias and myalgias are part of the picture, the syndrome is known as desert rheumatism. Skin rashes (erythema multiforme or erythema nodosum)
are also sometimes seen. Radiographic abnormalities can include infiltrates, hilar adenopathy, and pleural effusions. Symptoms generally last 2 to 3 weeks, but some patients can have persistent symptoms lasting weeks to months.
are also sometimes seen. Radiographic abnormalities can include infiltrates, hilar adenopathy, and pleural effusions. Symptoms generally last 2 to 3 weeks, but some patients can have persistent symptoms lasting weeks to months.
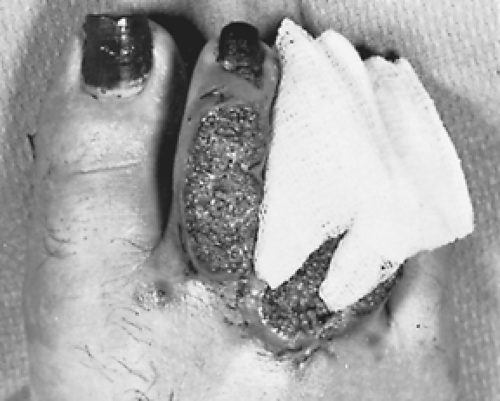 Figure 92-8. Characteristic skin lesions of blastomycosis on the dorsum of toes. (From Takaro T. Thoracic actinomycetic and mycotic infections. In Goldsmith HS, ed. Practice of Surgery. New York: Harper & Row, 1978. With permission.) |
As the acute process resolves, the infiltrates condense, leaving residual pulmonary nodules in approximately 5% of patients (Fig. 92-12). These are usually solitary and do not cause symptoms. In endemic areas they may be the most common cause of solitary pulmonary nodules. Unfortunately they can be very difficult to distinguish from a new presentation of lung cancer, especially if there is no previous chest x-ray for comparison. Coccidioides nodules are often moderately metabolically active on PET scanning, with standardized uptake values at or above the values considered the upper limit of normal for benign disease, but they can be extremely hypermetabolic as well. Even years later, these nodules often have viable C. immitis spherules within them, making transthoracic fine-needle aspiration a valuable tool in discriminating between Coccidioides nodules and cancer.
With the increasing population of retirees spending winters in southern Arizona and summers in more northerly locations, these types of nodules can create significant diagnostic dilemmas for physicians outside of endemic areas. If a nodule grows over serial computed tomography (CT) scans and either cannot be biopsied or biopsy is nondiagnostic, surgical excision is often warranted for diagnosis.
With the increasing population of retirees spending winters in southern Arizona and summers in more northerly locations, these types of nodules can create significant diagnostic dilemmas for physicians outside of endemic areas. If a nodule grows over serial computed tomography (CT) scans and either cannot be biopsied or biopsy is nondiagnostic, surgical excision is often warranted for diagnosis.
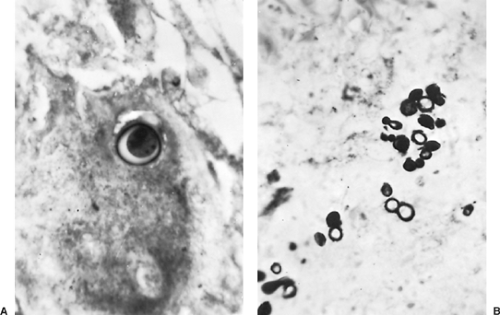 Figure 92-9. Blastomyces dermatitidis in resected lung tissue. A: Single thick-walled yeast form with refractile cell wall. B: Multiple yeast forms with single bud (periodic acid–Schiff stain, original magnification ×1,100). (From Takaro T. Thoracic actinomycetic and mycotic infections. In Goldsmith HS, ed. Practice of Surgery. New York: Harper & Row, 1978. With permission.) |
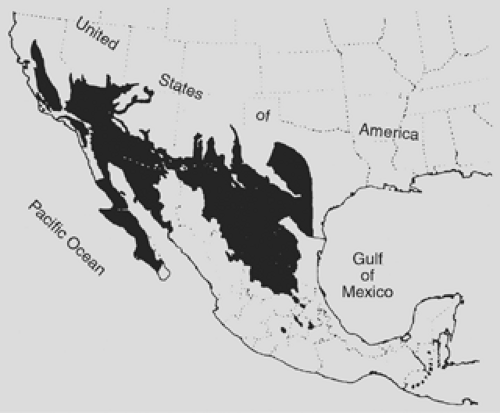 Figure 92-10. Map of the United States and Mexico showing the area of endemicity of coccidiodomycosis. (From Ajello CL, ed. Coccidioidomycosis. Tucson: University of Arizona Press, 1967. With permission.) |
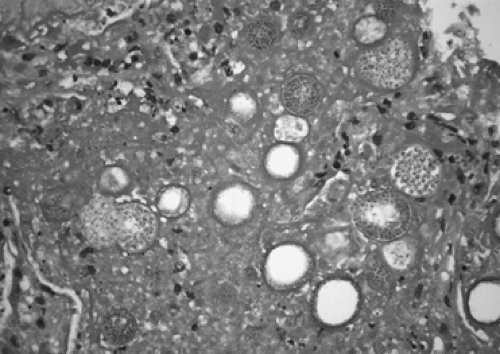 Figure 92-11. Coccidioides immitis in a resected lung nodule showing multiple spherules containing endospores. |
The other common sequela of acute infection is the cavitary lesion (Fig. 92-13). This is also seen in approximately 5% of patients after acute infection. Cavities are typical thin-walled, solitary, and subpleural in location. Most are asymptomatic and many, especially smaller cavities, will regress over a several-year time period. Cavities may cause chronic cough, chest pain, or hemoptysis, rarely massive hemoptysis. Fungus balls occasionally develop inside established cavities. Again, surgical excision can be required to differentiate these lesions from a cavitating lung cancer. Occasionally significant hemoptysis or progressive growth of the cavity will lead to surgical resection for cavities known to be benign in origin. In these cases, anatomic resections are often required, either segmentectomy or lobectomy, as the lung tissue surrounding the cavity can be acutely inflamed and edematous or densely fibrotic, making wedge resection technically challenging. Very rarely, a cavity can rupture into the pleural space, creating a hydropneumothorax. If the etiology of
the hydropneumothorax is recognized, it is valuable not only to drain the pleural space but also to remove the offending cavity.
the hydropneumothorax is recognized, it is valuable not only to drain the pleural space but also to remove the offending cavity.
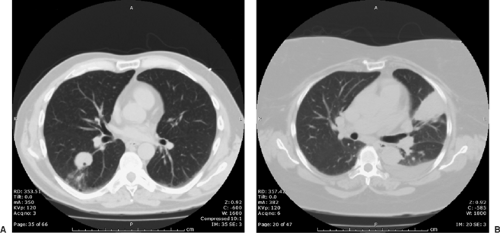 Figure 92-12. Solitary pulmonary nodule due to Coccidioides immitis. A: Solitary nodule. B: Solitary nodule with acute active infection, associated pleural effusion, and seropositivity.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|