Myasthenia Gravis
David S. Younger
This chapter provides an overview of the pathogenesis, diagnosis, and treatment of myasthenia gravis (MG).
Historical Aspects
The history of MG is controversial.39,40 The first description of a patient with MG appeared in 1644 in correspondence from colonial Jamestown, Virginia, pertaining to Indian Chief Opechankanough, according to Marsteller.15 In 1685, Sir Thomas Willis described a patient with bulbar symptoms that could have been psychogenic. The clinical syndrome of MG was identified by Wilks,36 Erb,6 and Goldflam.8 In 1895, Jolly12 named the disease myasthenia gravis pseudoparalytica. By 1900, Campbell and Bramwell3 had reported 60 cases. The efficacy of physostigmine was shown by Walker in 1934.32 One year later, he suggested the chemical nature of neuromuscular transmission at motor endplates.33 In 1941, Harvey and Masland10 summarized the salient electrophysiologic features of MG. In the same year, Blalock and coworkers1 and later Keynes14 described transsternal thymectomy in MG, including as complete a removal of the gland as possible whether or not a tumor was suspected preoperatively.
In 1960, an autoimmune cause of MG was suggested by Simpson25 and by Nastuk and colleagues.18 However, the immunologic basis of MG awaited basic understanding of acetylcholine (ACh) release at motor endplates, as subsequently described by Katz and Miledi.13 In 1973, Patrick and Lindstrom20 injected rabbits with acetylcholine receptors (AChRs) from the electric organ of eels, intending to make antireceptor antibodies and see whether these antibodies would block the function of AChR in intact electric organ cells. The antibodies did block, and the immunized rabbits became paralyzed and died. Experimental autoimmune myasthenia gravis (EAMG), so named, resulted from the autoimmune attack against native AChRs. Fambrough and associates7 applied alpha-bungarotoxin to motor-point biopsy samples from patients with MG and found a marked reduction in AChRs, averaging 20% of controls. Within the next several years, investigators reproduced the essential clinical and morphologic correlates of human MG in animals by passive transfer of human myasthenic serum and AChR-specific monoclonal antibodies.
Pathophysiology
Acetylcholine Receptor Module
The 1980s and 1990s witnessed spectacular progress in the understanding of the microstructure, physiology, and molecular composition of the nicotinic AChR. This, in turn, has been
applied to the clinical problem of MG. The AChR is a gated channel and intrinsic membrane glycoprotein of molecular weight 290,000, composed of five subunits. Distinct but related genes encode the individual subunits, and complementary DNA for each has been cloned, showing remarkable homology.
applied to the clinical problem of MG. The AChR is a gated channel and intrinsic membrane glycoprotein of molecular weight 290,000, composed of five subunits. Distinct but related genes encode the individual subunits, and complementary DNA for each has been cloned, showing remarkable homology.
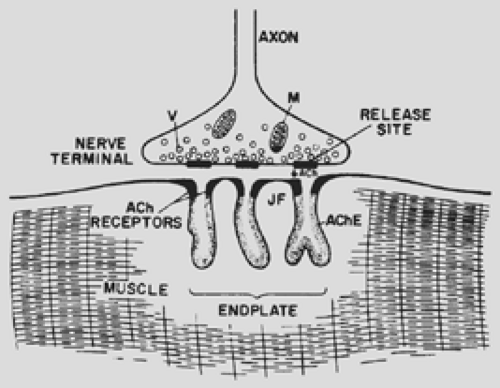 Figure 180-1. Neuromuscular junction. Vesicles (V) release their acetylcholine (ACh) contents at specialized release sites. After crossing the narrow synaptic space (arrow), ACh reaches the ACh receptors, which are most densely situated at the peaks of the junctional folds (JF). Acetylcholinesterase (AChE) in the clefts rapidly hydrolyzes the ACh. M, mitochondria. (From Drachman DB. Myasthenia gravis. Part 1. N Engl J Med 1978;298:136, 1978. With permission.) |
Neuromuscular Junction Physiology
The mechanisms of the end plate current (EPC) and end plate potential (EPP) have been elucidated by noise analysis and patch or voltage clamping. In response to an incoming nerve action potential, highly localized regions of the nerve terminal (Fig. 180-1) release approximately 200 quantal packets, each containing 6 to 10,000 molecules of ACh. Binding of ACh to specific sites on the AChR results in the transient openings of the AChR channel, allowing a net influx of Na+ ions and thus producing the depolarizing potential. The circular arrangement of the five subunits delineates a 2.5-nm channel whose narrowest point is 0.65 nm in diameter. Each subunit contains four membrane-spanning alpha helices termed M1 to M4, with the M2 segment effectively lining the channel. More quantal packets of ACh are released into the synaptic cleft, and more receptor channels are present than are needed to depolarize the muscle fiber to threshold. This creates a “safety factor.” Even when the number of receptors is reduced experimentally, the EPP does not fall below the threshold needed to generate a muscle action potential. The autoimmune attack on AChRs, which leads to the reduction in the number of functioning receptors at myasthenic end plates, also leads to many EPPs falling below threshold. This translates into muscle weakness and fatigue, particularly with repetitive or sustained contraction.
Autoimmune Etiopathogenesis
The hypothesis that MG originated in the thymus gland, as first suggested by Weigert35 in 1901, was difficult to prove and still remains speculative. The presumed initial event in the pathogenesis of myasthenia is loss of self-tolerance, a process attributed to the thymus gland for several intuitive reasons. The thymus contains all of the elements theoretically necessary for the activation of AChR-specific T cells. They include local antigen-presenting cells (APCs) or, in this case, myoid cells, which take up AChR derived from myoid cells, process it, and then express AChR-derived peptide fragments on their surface in the context of the trimolecular complex. The latter includes the APC, AChR-specific antigen linked to the major histocompatibility complex class II molecules, and AChR-specific reactive T cells. Although myoid cells are equally abundant in normal and myasthenic thymuses, hyperplastic glands contain many more myoid cells than atrophic glands. AChR-specific T cells are enriched in myasthenic thymus glands with and without epithelial cell tumors (thymomas). Whether the AChR-specific T cells in the myasthenic thymus always resided there or return after a sojourn in the peripheral immune system is not known. The peripheral blood of patients with MG contains an enhanced portion of these autoreactive T cells, which are capable of recruiting AChR-responsive B cells for the production of pathogenic anti-AChR antibodies. In actively induced or passively transferred EAMG, where the myasthenic process is initiated outside the gland, germinal centers are not observed. Finally, transplantation of myasthenic thymus fragments into mice with severe combined immunodeficiency results in the production of pathogenic mouse antibodies.
Diagnosis
Nosology and Classification
The term myasthenia has been used interchangeably for the acquired autoimmune form of the disease, and the term myasthenic has generally been used for other syndromes of the neuromuscular junction. Similarly, the classification of MG has been difficult. Early attempts emphasized duration of symptoms because it was believed that the disorder might be progressive. Younger and colleagues37,38 emphasized indices of maximal severity, functional status, and response to therapy, prompting more exact nosology, classification, and comparative methods of analysis of patient outcome.
Clinical Aspects
The clinical diagnosis of autoimmune acquired MG is made by recognizing a pattern of weakness that has the features of fluctuation and variability over the course of a day, months, or years, leading to perceptible exacerbations and remissions. The distribution of weakness is characteristic, affecting ocular, facial, oropharyngeal, and limb muscles. The useful Osserman and Genkins classification is listed in Table 180-1. The diagnosis is confirmed by unequivocal and reproducible improvement after intravenous administration of edrophonium chloride, a rapidly acting anticholinesterase drug. Formal diagnosis is bolstered by eliciting a decremental response to repetitive motor nerve stimulation as well as the detection of AChR antibodies in the serum. Selective involvement of limb and respiratory muscles, sparing ocular or oropharyngeal muscles, is rarely if ever encountered.
Table 180-1 Osserman and Genkins Classification | ||
|---|---|---|
|
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


