Chapter 42 Multiple Endocrine Neoplasia Syndromes
Multiple Endocrine Neoplasia Type 1
Genetic Studies and Pathogenesis
The MEN1 tumor suppressor gene, whose mutations are responsible for the MEN1 syndrome, was originally mapped to human chromosome 11q13 by genetic linkage studies and tumor DNA deletion mapping, and was identified by positional cloning in 1997.1 MEN1 is a putative tumor suppressor gene, whose protein product likely has diverse actions that confer a negative influence or brake on cellular growth and proliferation, so that loss of its function results in unregulated cell growth or neoplastic transformation. As a tumor suppressor gene, development of MEN1 requires two genetic hits involving both allelic copies of the gene to result in loss of function. The first mutation is inherited in the germline and is present in every cell; the second somatic mutation occurs in an individual cell of an involved target tissue and results in tumor formation. According to the two-hit model of tumorigenesis, the first event is a mutation inherited in the germline that confers susceptibility to neoplastic change in the involved tissues. Elimination of the remaining functional copy of the gene in a single cell through a chance somatic mutational event, or second hit (such as a gene deletion), results in clonal expansion and cancer development. The occurrence of individual second hits in several target organ cells explains the multifocal involvement characteristically observed in affected endocrine tissues. Somatic mutations in the MEN1 gene occur frequently in sporadic parathyroid adenomas, insulinomas and gastrinoma, pituitary tumors, and bronchial carcinoids, indicating that loss of the MEN1 gene contributes to the development of a subset of nonhereditary endocrine tumors.
The MEN1 gene consists of 10 exons (9 coding and 1 untranslated) and encodes a 610–amino acid protein termed menin.1 Menin is ubiquitously expressed in endocrine and nonendocrine tissues. The menin protein sequence is highly conserved evolutionarily, with the murine Men1 gene demonstrating 98% homology. Knockout of both Men1 alleles in mice results in embryonic lethality, demonstrating that menin is essential for early development and has a broader role in the regulation of cell growth that is not limited to the endocrine tissues affected in MEN1. An excellent mouse animal model for the study of MEN1 tumorigenesis exists. Heterozygous Men1± mice demonstrate somatic loss of the wild-type Men1 allele in tumors2 and develop a constellation of endocrine tumors remarkably similar to the human MEN1 syndrome.
Menin is now known to have diverse influences in the regulation of gene transcription, cell cycle progression, apoptotic pathways, DNA processing and repair, cytoskeletal integrity, and genome stability. Menin is predominantly a nuclear protein that binds to Jun, a member of the AP-1 transcription factor family, and represses JunD-mediated transcription.3 In addition, menin has been shown to interact physically with a diverse variety of other proteins that comprise transcription factors, DNA-processing factors, DNA repair proteins, and cytoskeletal proteins (e.g., Smad3, NF-κB, nm23, Pem, FANCD2, RPA2, ASK). Overexpression of menin has been shown to diminish the tumorigenic phenotype of Ras-transformed NIH-3T3 cells, consistent with its putative tumor suppressor function. In addition, studies have suggested a possible role for menin in repressing telomerase activity in somatic cells, perhaps explaining in part its tumor suppressor properties. Although there is now rapidly expanding evidence regarding diverse menin interactions and influences on a variety of cellular functions and pathways, the full complexity of the tumor suppressor role of menin in MEN1 and sporadic endocrine tumorigenesis has not yet provided a comprehensive understanding of the mechanisms involved.
To date, more than 1330 independent mutations have been identified in the MEN1 gene.4 MEN1 mutations may be nonsense, missense, frameshift, deletions, and RNA splicing defects. These diverse mutations may occur throughout the coding sequence and intron-exon junctions of the gene, as demonstrated in a representative study of MEN1 germline mutations (Fig. 42-1).5 Approximately two thirds of the reported mutations in the MEN1 gene result in premature termination of translation and truncation of the C-terminal portion of the menin protein. No clear genotype-phenotype correlation has been established for MEN1, although phenotypic variants (isolated hyperparathyroidism, frequent prolactinomas) have been described.6
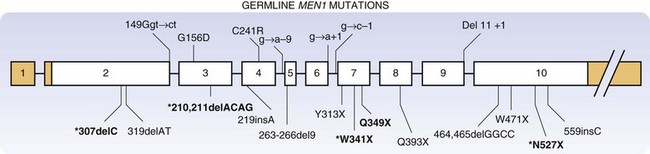
Because no clear correlation has been found between specific mutations in the MEN1 gene and phenotype of the affected patients, the use of genetic testing for the prediction of malignant potential and prognosis is not possible at present.7 When an index-suspected MEN1 patient is diagnosed clinically, genetic evaluation of that patient and first-degree relatives should be performed. Presymptomatic individuals who test positive for an MEN1 mutation should then undergo more frequent and intensive biochemical testing, with special emphasis on the potentially malignant pancreaticoduodenal and intrathoracic tumors. Close observation and frequent surveillance of patients with MEN1 mutations allows for much earlier detection of biochemical abnormalities associated with neoplasia.8 Conversely, a negative genetic test result in a patient from a family whose specific mutation is known would obviate further lifelong screening or testing, with the associated costs and psychological impact.
Clinical Features and Management
The clinical manifestations of patients with MEN1 depend on the endocrine tissue involved, specific hormone overproduced, or local mass effect and malignant progression of the neoplasm. Previously, complications related to hormone excess, such as severe ulcer disease or hypoglycemia, were the most frequent presenting complaints. At present, the principal cause of mortality in patients with MEN1 is malignant progression of duodenopancreatic neuroendocrine cancers or intrathoracic malignant carcinoids. Current recommendations indicate that annual biochemical testing, including determination of pancreatic polypeptide, gastrin, glucagon, and chromogranin A levels, should begin at approximately age 15 to 20 years in presymptomatic affected individuals.9 It is now recommended to begin biochemical screening for pituitary adenomas in carriers of MEN1 mutations at 5 years of age. Some have recommended screening for hyperparathyroidism as early as the age of 8 years in MEN1 mutation carriers, but the peak incidence of clinically apparent HPT is in the late second or early third decade. A summary of our recommendations for the clinical and biochemical surveillance of patients with MEN1 is shown in Figure 42-2.
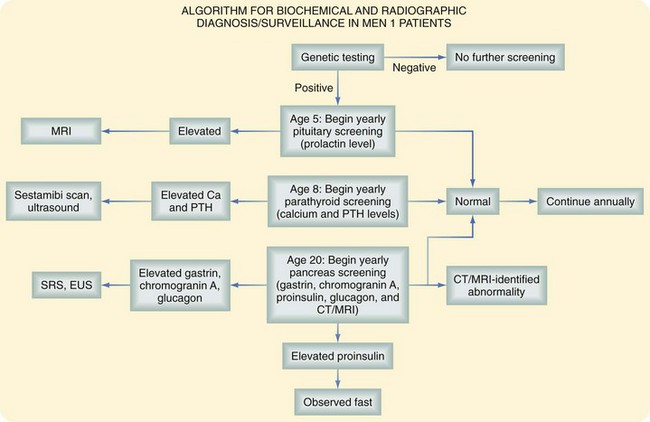
FIGURE 42-2 Clinical and biochemical surveillance in patients with MEN1.
(From Whaley JG, Lairmore TC: Multiple endocrine neoplasia type 1: Current diagnosis and management. In Morita SY, Dackiw APB, Zeiger MA [eds]: McGraw-Hill’s manual of endocrine surgery, New York, 2009, McGraw-Hill, pp 334–347.)
Parathyroid Glands
The most common endocrine abnormality (>98% affected individuals) in MEN1 is multiglandular parathyroid tumors. The parathyroid tumors in MEN1 are clonal, resulting from inactivation of both alleles of the MEN1 tumor suppressor gene by two separate events, and are therefore multiple adenomas from a strict genetic standpoint.10 In contrast, fewer than 15% of patients with sporadic primary hyperparathyroidism have multiglandular involvement. The typical enlargement of parathyroid glands in MEN1 patients is asymmetrical at any point in the intervention (Fig. 42-3).
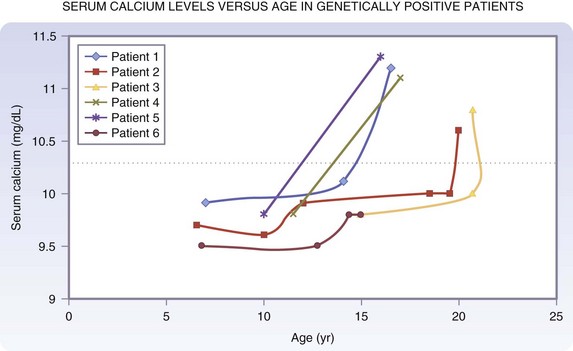
Hypercalcemia is usually the first biochemical abnormality detected in patients with MEN1, and may precede the clinical onset of a pancreatic NET or pituitary neoplasm by several years. Asymptomatic hypercalcemia may be present in many patients over a long period of observation. Renal lithiasis and skeletal complications of hyperparathyroidism occur but are uncommon. In general, hyperparathyroidism in patients with MEN1 has an earlier age of onset and usually causes a milder hypercalcemia than that observed in primary sporadic hyperparathyroidism. Demonstrating an elevated serum calcium level in association with an inappropriately elevated parathyroid hormone level establishes the diagnosis. Patients with MEN1 typically have a markedly elevated 24-hour urine calcium excretion. When prospective biochemical screening of known genetically positive individuals is performed, the onset of hypercalcemia in patients with an MEN1 mutation occurs as early as 11 to 14 years of age (Fig. 42-4).
Debate continues regarding the optimal surgical procedure for hyperparathyroidism in patients with MEN1. The incidence of recurrent hyperparathyroidism following any surgical treatment for the multiglandular disease in MEN1 is approximately 30% to 40% 5 years postsurgery, reflecting the genetic first-hit predisposition in every parathyroid cell. Recurrences are related to how long the patient is followed and the performance of thymectomy.11 A potential advantage of total parathyroidectomy and heterotopic autotransplantation to the forearm is the ability to manage recurrent hyperparathyroidism, if it develops, by excision of a portion of the grafted parathyroid tissue under local anesthesia, obviating the morbidity of repeat neck exploration. Subtotal resection is believed by many to achieve equivalent results without a purported higher risk of permanent hypocalcemia from autograft failure.12 Currently, both treatments appear to provide essentially equivalent results, and the answer to this question awaits a randomized, prospective clinical trial. Delayed transplantation of cryopreserved autologous parathyroid tissue can salvage a proportion of patients with permanent postoperative hypocalcemia following either procedure. In one study,13 approximately 60% of delayed, cryopreserved parathyroid autografts showed evidence of graft function based on venous parathyroid hormone (PTH) gradients between the grafted and nongrafted arms; 40% of autografts achieved full competency without supplements.
Pancreas and Duodenum
Endoscopic ultrasonography (EUS) is an excellent imaging modality for NETs of the pancreas and duodenum but is dependent on availability and operator skill in selected centers. In 1992, Rosch and colleagues14 have reported a sensitivity of 82% and specificity of 95% of EUS in detecting pancreatic NETs that had not been localized by transabdominal ultrasound or CT. However, the detection rate decreases with distal progression along the pancreatic tail, likely because of increased distance from the gastric or duodenal lumen. EUS is an effective and relatively noninvasive localizing test (after initial CT), but is dependent on the skill and experience of the operator and availability.
Somatostatin receptor scintigraphy (SRS) may also be used to localize these tumors. In the subpopulation of MEN1 patients, one study found that the specificity and positive predictive value of SRS for pancreatic tumors are 25% and 100%, respectively, whereas specificity and positive predictive value of SRS for duodenal gastrinomas are 72% and 100%, respectively.15 In this study, all pancreatic NETs were detected by EUS or SRS.
Finally, selective pancreatic arteriography with provocative stimulation by selected secretagogues and measurement of increment hormone secretion in the hepatic vein is an invasive test, but may be the most accurate single localizing study.16 This test provides regional localization of functional tumors (e.g., insulinoma, gastrinoma) within the pancreas and is especially useful for patients with MEN1 who characteristically have multiple tumors.
Gastrinomas that develop in patients with MEN1 are usually malignant (≈80%), as indicated by the presence of regional lymph node or distant metastases. Gastrinomas were previously believed to be located predominantly in the head of the pancreas within the gastrinoma triangle. More recent data have suggested that gastrinomas in patients with MEN1 occur most frequently within the wall of the duodenum (Fig. 42-5). Because of the small size of these neoplasms, the primary gastrinoma may not be localized preoperatively by CT scanning or angiography. Endoscopic ultrasound has been used successfully to localize gastrinomas within the wall of the duodenum or head of the pancreas. There is controversy about the development of primary gastrinoma within lymph nodes. Although occasional patients have been biochemically cured after resection of gastrinoma within lymph nodes, it is unclear whether an occult gastrinoma primary was missed within the pancreas or wall of the duodenum.
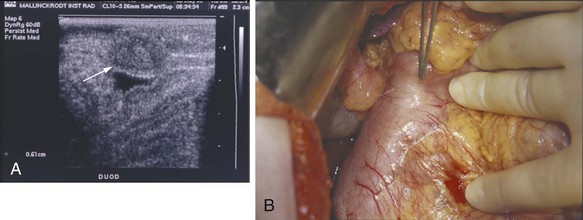
The value of surgical resection for intended cure of gastrinoma in patients with MEN1 is controversial. Although most evidence indicates that patients with ZES and MEN1 rarely demonstrate long-term biochemical cure following operation,17 localized resection of a potentially malignant NET is indicated in an attempt to control the tumoral process and prevent subsequent malignant dissemination. The recognition that primary gastrinomas occur frequently in the duodenal wall, combined with efforts to perform an extensive regional lymphadenectomy or even pancreaticoduodenectomy, may improve the success rate of surgery for ZES in the setting of MEN1.18 Newer strategies, including rapid intraoperative gastrin determinations, are also being used in the surgical management of gastrinoma. Total gastrectomy is rarely indicated for patients with gastrinoma because medical therapy effectively prevents most of the symptoms or complications resulting from the acid hypersecretion. The availability of PPIs allows effective medical therapy for patients with unresectable gastrinoma or extensive metastatic disease. However, careful observation of the stomach by repeated endoscopy is necessary because long-term administration of PPIs to patients with MEN1 and ZES has been associated with the development of gastric carcinoid tumors. Patients with primary hyperparathyroidism should undergo parathyroidectomy because normalization of the serum calcium level improves the ZES.19
There is no ideal medical therapy for insulinoma; therefore, the preferred treatment is accurate localization and surgical resection of the functioning tumor to correct life-threatening hyperinsulinemia. Patients with MEN1 characteristically develop multiple NETs, which may complicate identification of the specific functional tumor responsible for the hyperinsulinism. Preoperative regional localization of the functioning tumor within the pancreas may be provided by selective catheterization of the arteries supplying the pancreas, followed by injection of an insulin secretagogue (calcium gluconate) and measurement of insulin gradients in the hepatic veins. The operative approach includes complete mobilization of the pancreas and careful examination of the gland by inspection and palpation. Intraoperative ultrasound is essential for the identification of small tumors, especially within the pancreatic head or uncinate process.20 Small benign insulinomas are amenable to enucleation. Partial pancreatectomy may be required for multiple or potentially malignant tumors.16 If the insulinoma is not identified, despite an exhaustive intraoperative search, blind subtotal pancreatectomy is not recommended. Approximately 10% of insulinomas occurring in patients with MEN1 are malignant. Patients with malignant insulinoma and disseminated metastases may respond to treatment with streptozotocin and some control of hypoglycemia may be achieved by the administration of diazoxide or octreotide.
The involvement of NETs within the pancreas of patients with MEN1 is characteristically multifocal (Fig. 42-6). Controversy exists regarding the optimal timing and most appropriate operation to perform for NETs of the pancreas and duodenum in patients with MEN1. The controversy reflects uncertainty about the natural history of small, potentially benign or nonfunctional tumors, which must be weighed against the risks of early and/or repeat major pancreatic interventions carrying a significant risk of morbidity. Some are reluctant to advocate routine or early pancreatic exploration in young, otherwise healthy patients for small nonfunctional tumors, which might be clinically insignificant. On the other hand, these tumors have a malignant potential, and delay in diagnosis and effective treatment carry the risk of the development of local or distant metastasis. It is obviously desirable to intervene early to prevent malignant dissemination while minimizing morbidity and mortality (from cancer or surgery). Complicating factors include the lack of genotype-phenotype correlation in MEN1, which might otherwise allow genetic stratification of those at higher risk of malignant progression, and failure of recent studies to identify a clear relationship between the size of the tumor and risk of regional lymph node or distant metastases.21
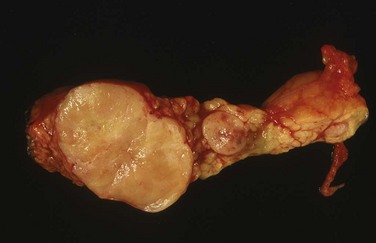
FIGURE 42-6 Multiple neuroendocrine tumors of the pancreas in a distal pancreatectomy specimen from a patient with MEN1.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


