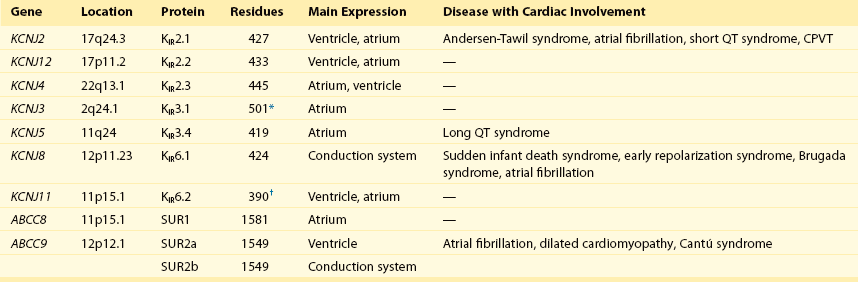
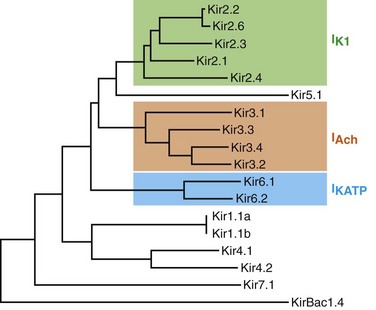
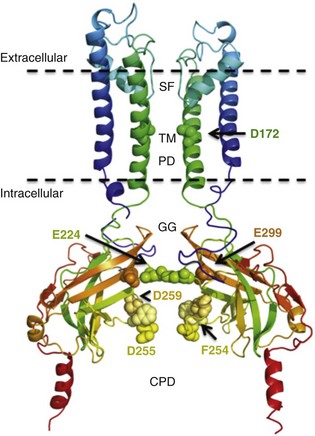
13 The description in 1949 by Katz1 of the inward rectifier current that he considered “an interesting phenomenon, but difficult to explain” in muscle can be regarded as the scientific birth of inward rectifier currents. Inward rectifier potassium currents (KIR) are characterized by their key feature of allowing more inward than outward potassium flow at equivalent membrane potentials negative and positive of the potassium equilibrium potential. Inward rectifier channels are formed by tetramerization of the underlying pore-forming KIR channel proteins. Ion channel regulation takes place mainly via the intracellularly located amino- and carboxyl-terminal regions of the proteins. Functionally, inward rectifiers define, for a large part, the membrane potential of nonexcitable cells and the resting membrane potential of excitable cells. The eukaryotic inward rectifier family is encoded by the KCNJ genes and includes seven subfamilies (KIR1-7) (Figure 13-1). Each protein consists of two transmembrane domains, a short pore loop that harbors the potassium selectivity filter (GYG) and intracellularly located N- and C-termini (Figure 13-2). Between subfamilies, sequence homology is as high as 40%, whereas sequence identity rises to approximately 60% within subfamilies. Each of the subfamily gene products display their own characteristics, some having strong and others weak rectifying potential and some responding to metabolic stimuli or neurotransmitters directly. Individual family members are involved in many physiologic processes such as cardiac and neural excitability, insulin release, vascular tone, and potassium homeostasis.2 Members of three different subfamilies are main contributors to cardiac electrophysiology (Table 13-1). The classical inward rectifier current (IK1) channel is formed by KIR2.x proteins and stands at the basis of the resting membrane potential in working cardiomyocytes; the acetylcholine (ACh) responsive rectifiers are built of KIR3.x proteins and play an eminent role in heart rate modulation, whereas the adenosine triphosphate (ATP)-sensitive channels are formed by octamers of KIR6.x and the regulatory sulfonylurea receptor (SUR) proteins and are responsible for coupling the metabolic state of the vulnerable cardiomyocyte to its electrical activity.3,4 Table 13-1 Human Cardiac Inward Rectifier Genes, Expression, and Associated Cardiac Disease CPVT, Catecholaminergic polymorphic ventricular tachycardia. *Longest isoform. Shorter isoforms of 235, 253, and 308 amino acids have been reported. †Longest isoform. Shorter isoform of 303 amino acids has been reported. Figure 13-1 Cladogram of the human KCNJ gene family. Amino acid sequences were aligned by a Clustal-W algorithm, phylogeny was determined by the neighbor-joining method. Bacterial KIRBac1.4 was used as outgroup. Subfamilies relevant for cardiac electrophysiology are color-coded: classical inward rectifier genes (green), acetylcholine-regulated channel genes (orange), and ATP-sensitive inward rectifier genes (blue). Figure 13-2 Structure model of KIR2.1 ion channel. Negatively charged amino acid residues (D172, E224, F254, D255, and D259) involved in polyamide-mediated rectification face the transmembrane and cytoplasmic pore regions, respectively. CPD, cytoplasmic pore domain; GG, G-gate; SF, selectivity filter; TMPD, transmembrane pore domain. (Used with permission from Van der Heyden MA, Sánchez-Chapula JA: Toward specific cardiac IK1 modulators for in vivo application: old drugs point the way. Heart Rhythm 8:1076–1080, 2011.) Each monomer comprises a transmembrane (TM) and a cytoplasmic part. The transmembrane domain (TMD) consists of the two TM helices, the pore helix positioned between these, and a slide helix located in front of TM1. The cytoplasmic pore domain (CPD) is compiled by interaction of the amino- and carboxyl-termini. In the resulting channel, the four TM2 (inner) helices of each monomer face the central water-filled pore in the TMD, with the potassium selectivity filter (SF) positioned in close proximity to the cellular exterior (see Figure 13-2). It is believed that ion conduction may be regulated by two gates in series: one formed by the inner helices (TM2) of the TMD at the helix-bundle crossing and the other by the G loop at the apex of the CPD.2 Tetramerization of CPDs creates an extended pore region into the cytoplasm, separated from the TM pore region by the G-loops. Polyamine block of inwardly rectifying potassium (KIR) channels underlies their key functional property of preferential conduction of inward K+ currents (inward rectification). Kinetic models describe polyamine block as a multistep process, incorporating sequentially linked “shallow” and “deep” binding steps of polyamines in the KIR pore. Structurally, these shallow and deep binding steps are conceptualized as an initial weakly voltage-dependent binding in the cytoplasmic domain of the channel, followed by a steeply voltage-dependent step in which spermine migrates to a stable binding site in the TM pore (inner cavity). Five negatively charged residues within the cytoplasmic (E224, D259, E299, F254, and D255) and one in the TM pore regions (D172) are involved in interacting with the positively charged polyamines.5 Functional cardiac ACh regulated inward rectifier current (IKACh) channels are formed by heterotetramers of KIR3.1 and KIR3.4 (also known as GIRK1 and GIRK4), encoded by the KCNJ3 and KCNJ5 genes, respectively. Expression is confined mainly to the atrium, and functional expression in the sinus and atrioventricular nodes is an important feature for heart rate and atrioventricular conduction regulation. Homotetramers of KIR3.1 or KIR3.4 produce less or no current when overexpressed in Xenopus oocytes.6 It was established that a stochiometry of 1 : 1 and an alternating position of KIR3.1 and KIR3.4 subunits within the channel produced most robust IKAch channel activity.2 The structural organization of the IKAch channel is similar to the classical IK1 channel. The TM2 domains face the water-filled pore and, confined by the G-gate, an extended pore region protrudes into the cytoplasm. A leucine residue within the C-terminus of KIR3.1 (L333) and KIR3.4 (L339) appears crucial for Gβγ-dependent activation. Interestingly, several domains involved in Gβγ-dependent activation are formed at the cytoplasmic-facing interfaces of the KIR3.1 and KIR3.4 interacting domains.2 Compared with IK1 and IKAch channels, an additional level of complexity is found in the ATP-sensitive rectifier current (IKATP) channels, because a tetramer of SUR proteins embraces the KIR-formed channel.7 Cardiac SUR protein–expressing genes ABCC8 and ABCC9 are members of the large ATP-binding cassette (ABC) superfamily that consists of 48 genes divided into seven subfamilies.8 SUR1 (ABCC8) and SUR2 (ABCC9) contain the typical ABC “core” that consists of two membrane domains (TMD1 and TMD2) of six TM helices, each separated by two cytoplasmic nucleotide–binding domains (NBD1 and NBD2). Furthermore, the proteins contain an additional N-terminal–located membrane domain consisting of five TM helices (TMD0) that are linked to the “core” domain by the L0 linker. Interaction with the KIR6.x-formed pore is established mainly by the TMD0 and L0 domains. Nucleotide binding drives dimerization of NBD1 and NBD2, which results in subsequent rearrangements of TMD1 and TMD2 and alterations in IKATP channel gating. The pore-forming KIR6.x complex resembles the structures of KIR2.x and KIR3.x channels. Cardiac IKATP channels, depending on their location in the heart, consist of octaheteromers of four KIR6.2 (KCNJ11) and four SUR2a subunits (ventricle), KIR6.2 and SUR1 (atrium), or KIR6.1 (KCNJ8)/KIR6.2 and SUR2b (conduction system). In the working myocardium, KIR2.x-mediated IK1 sets the resting membrane potential close to the potassium reversal potential and contributes outward potassium current during repolarization. With respect to the heart, neonatal KIR2.1 knockout mice were bradycardic and displayed electrocardiogram (ECG) abnormalities such as lengthening of the PR (~50%) and QT (~50%) intervals. Isolated cardiomyocytes from KIR2.1 knockout animals displayed (1) no IK1; (2) action potential prolongation; and (3) spontaneous beating activity.9 In contrast, KIR2.2 knockout mice were viable and lived through adulthood without ECG abnormalities. However, their cardiomyocytes displayed a 50% reduction in IK1 densities. In humans, dominant negative KCNJ2 mutations are associated with Andersen-Tawil syndrome 1, characterized by periodic muscle paralysis, biventricular tachycardia and sometimes long QT times, and developmental bone abnormalities (see Table 13-1).10 Gain-of-function mutations in KCNJ2 have been associated with short QT syndrome, catecholaminergic polymorphic ventricular tachycardia, and congenital atrial fibrillation.11–13 Currently, no disease-causing mutations in KCNJ12 and KCNJ4 have been described. IKACh slows heart rate and atrioventricular conduction in response to muscarinic M2 receptor activation by ACh released from the vagus nerve. In this process, Gβγ directly interacts with and activates the channel that thereby antagonizes diastolic depolarization of the nodal cardiomyocytes. KCNJ3–/– mice were viable but showed a lack of carbachol-induced IKACh in atrial cardiomyocytes. Moreover, animals showed blunted responses to pharmacologic interventions directed at vagal-induced bradycardia.14 KCNJ5–/– mice15 displayed loss of cardiac IKAch. Mice were viable; in most studies their resting heart rate increased and animals showed a reduced negative chronotropic response after vagal stimulation. Furthermore, mice were unable to undergo rapid changes in heart frequency. In human heart function, KCNJ5 mutations have been associated with long QT syndrome, syncope, atrial fibrillation, and sudden cardiac death (see Table 13-1).16 No diseases related to mutations in KCNJ3 have been reported. KIR6.1 knockout mice die suddenly after the occurrence of ST elevation and subsequent third-degree atrioventricular block and cessation of heart rhythm, which may result from myocardial ischemia as a result of blunted coronary vasoregulation.17 The presence of KIR6.1 in the cardiac conduction system may provide another or additional mechanism of cardiac pathology and conduction disturbances in these animals. Furthermore, KIR6.1 knockout mice are more susceptible to endotoxin-mediated septic shock, resulting in ST elevation and cardiac death. In the hearts of KIR6.2 knockout mice, a number of stressors induce calcium overload, followed by short-term abnormalities in cardiac rhythm (early afterdepolarization, premature ventricular contractions, and sinus node regulation) and hemodynamics (increased left ventricular end-diastolic pressure).18,19 In the long term, cardiac maladaptive remodeling (exaggerated fibrosis and hypertrophy) finally results in impaired ejection fraction and congestive heart failure. SUR1 knockout mice are fertile and phenotypically normal but lack atrial IKATP.20 Furthermore, ABCC8 null mutation increases resistance to cardiac, but not neuronal, ischemia-reperfusion injury.21 Knock out of SUR2 resulted in loss of IKATP from all muscles, coronary vasospasms, increased blood pressure, and premature death from week 5 onward. Animals displayed slight cardiac hypertrophy and increased resistance to cardiac ischemia-reperfusion damage.22 In humans, a gain-of-function mutation in KCNJ8 has been associated with early repolarization and Brugada syndrome, both characterized by specific (J-wave) ECG abnormalities and atrial fibrillation.23–26 KCNJ8 loss of function has been associated with sudden infant death syndrome.27 ABCC9 mutations have been linked to atrial fibrillation, dilated cardiomyopathy, and Cantú syndrome, which causes cardiomegaly and pericardial effusion, among other effects (see Table 13-1).28–30 Thus far, KCNJ11 and ABCC8 mutations have not been associated with cardiac phenotypes. The most commonly used blockers to examine the physiologic roles of KIR channels in native cells and tissues are Ba2+ and Cs+. At micromolar concentration, Ba2+ blocks KIR channels with relative specificity. Externally applied Ba2+ suppresses KIR currents in a voltage-dependent manner and Ba2+ inhibits KIR channels more strongly as the membrane is hyperpolarized.2 However, experiments have shown that BaCl2 infusion in vivo produces strong deleterious effects—mainly skeletal, cardiac, and smooth muscle dysfunction.5 In the intact heart, IK1 blockers produce membrane depolarization (an effect that slows conduction velocity because of a voltage-dependent inactivation of Na+ channels), cause prolongation of the QT and QRS intervals on the surface ECG, and at higher concentrations cause ventricular ectopy and lethal ventricular arrhythmias. Conversely, it has been demonstrated that increasing inward rectifier currents stabilize reentrant arrhythmias. Recently, it has been shown that pharmacologic inhibition of inward rectifier currents by chloroquine induces antifibrillatory effects.31 Different antiarrhythmic (e.g., quinidine, amiodarone, propafenone, disopyramide) and noncardiac (e.g., terfenadine, astemizole) drugs at micromolar concentrations have been found to inhibit the classical cardiac IK1. However, little is known about their mechanisms of inhibition.32 Gambogic acid (GA) is a potent inducer of apoptosis that has been considered for anticancer therapy. At 10 µM, GA inhibited KIR2.1 current differentially during acute applications that produced 30% of inhibition or during prolonged exposures (up to 3 hours), which produced 70% of inhibition. The IC50 of KIR2.1 inhibition by GA during prolonged exposures was 27 nM. GA–induced inhibition is not caused by changes in biogenesis or trafficking to the plasma membrane. It is a lipophilic molecule (LogP ~6.77) that can insert itself into the lipid bilayer and thereby might perturb association of KIR2.1 with regulatory lipids and proteins that specifically facilitate the function of KIR2.1 channels. It has been found that pretreatment of HEK293 cells with GA shifted KIR2.1 and Kv2.1 channels from the Triton X-100–insoluble to the Triton X-100–soluble fraction. GA decreasing KIR2.1 activity is consistent with KIR2.1 being physiologically active in the context of Triton X-100–insoluble microdomains, but not of Triton X-100–soluble microdomains.33 A different drug, the cell-permeable dienone phenol triterpene celastrol, a neuroprotective compound that is also a slow-acting compound, decreased the rate of ion channel transport and caused a reduction of channel density on the cell surface. Muscarinic ACh receptors in the peripheral nervous system are found primarily on autonomic effector cells innervated by postganglionic parasympathetic nerves. In the heart, the muscarinic receptor M2 is the predominant subtype, which is coupled to the Gi/o protein family. IKACh channels can be activated by muscarinic receptor agonists like cholinomimetic choline esters (ACh, methacholine, carbachol, and bethanechol). The muscarinic receptor antagonist alkaloids such as atropine and synthetic and semisynthetic compounds prevented the effects of ACh by blocking its binding to muscarinic receptors.34
Molecular Regulation of Cardiac Inward Rectifier Potassium Channels by Pharmacologic Agents
Background
Inward Rectifier Channel Proteins are Encoded by KCNJ Genes
KIR2.x Proteins Constitute the Classical Inward Rectifier Current
Cardiac Acetylcholine Responsive Inward Rectifiers are Formed by KIR3.1 and KIR3.4 Heteromers
IKATP Channels Are Octaheteromers of Four KIR6.x and Four SURX Channel Proteins
Cardiac Role of Inward Rectifier Channels
KIR2.x Channels Set the Resting Membrane Potential and Contribute to Repolarization
IKACh Channels and Heart Rate Regulation
IKATP Channels Confer Metabolic Status to Electrical Properties
Pharmacology of KIR Channels
Effects of Drugs on KIR Channels
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Molecular Regulation of Cardiac Inward Rectifier Potassium Channels by Pharmacologic Agents
