Fig. 7.1.
Molecular mechanisms of detection avoidance during interaction of Histoplasma yeasts with macrophages. Adherence and phagocytosis of yeasts into macrophages is mediated by cell-wall-localized Hsp60 binding to CD18-family receptors (e.g., CR3) in the macrophage plasma membrane. At the same time, Histoplasma yeasts avoid detection by the macrophage β-glucan receptor Dectin-1 by concealing fungal β-glucans beneath an α-glucan polysaccharide layer in the yeast cell wall. In addition, macrophage TLR2 receptors do not detect undefined TLR2 PAMPs present in the yeast cell wall
Binding to complement receptors for adherence also triggers internalization of yeasts into macrophages. Intracellular residence in macrophages enhances the virulence of Histoplasma by protecting yeast cells from pulmonary collectins, specifically surfactant proteins A (SP-A) and D (SP-D) (McCormack et al. 2003). Recent work has shown that SP-A and SP-D have direct fungicidal activity on Histoplasma by permeabilizing the yeast cells. SP-A knockout mice have increased susceptibility to Histoplasma infection in vivo (McCormack et al. 2003). This suggests that phagocytic uptake is not only tolerated by Histoplasma but important for full virulence because it provides a shelter from soluble host defense molecules. Unlike Fc-mediated phagocytosis of antibody-opsonized particles, complement receptor-mediated phagocytosis is generally noninflammatory unless accompanied by other stimulating signals (Aderem 2003).
B. Concealment of Pro-inflammatory Fungal Patterns
To reduce potential pro-inflammatory host responses, Histoplasma yeasts minimize cell wall β-glucan detection by concealing it beneath a layer of α-glucan. Macrophage-expressed Dectin-1 is a critical pattern recognition receptor (PRR) for detection of fungal cell wall β-glucan and stimulates the antifungal responses of phagocytes (Goodridge et al. 2009; Drummond and Brown 2011). Dectin-1 detection of Histoplasma yeasts is necessary for pro-inflammatory cytokine production (Rappleye et al. 2007; Lin et al. 2010). However, most Histoplasma species synthesize α-linked glucan (α-glucan) in addition to β-glucans, and these two polysaccharides are organized in the yeast cell wall such that the β-glucans are concealed beneath the α-glucan layer (Rappleye et al. 2007). Functionally, production of the yeast-specific α-glucan polysaccharide effectively masks the normally immunostimulatory cell wall β-glucans (Fig. 7.1). Yeast cells unable to synthesize α-glucan are readily detected by Dectin-1 and these yeasts are severely attenuated in vivo (Rappleye et al. 2004, 2007).
Synthesis of α-glucan requires the function of an α-(1,3)-glucan synthase (Ags1) as well as an α-(1,4)-amylase (Amy1) (Rappleye et al. 2004; Marion et al. 2006). One species of Histoplasma (the North American type 2; NAm 2) lacks detectable α-glucan yet remains virulent. This species appears to express an alternate means of preventing β-glucan detection through an unknown mechanism (Edwards et al. 2011a).
Thus, the α-glucan polysaccharide produced by most species of Histoplasma facilitates stealthy entry of Histoplasma yeast into macrophages, thereby enhancing yeast survival.
In addition to preventing Dectin-1 detection, Histoplasma yeast cells do not trigger Toll-like receptors (TLRs) expressed on the macrophage surface. The most relevant TLRs that recognize fungal pathogen-associated molecular patterns (PAMPs) are TLR2 and TLR4. One possibility is that Histoplasma yeasts lack TLR ligands altogether. Consistent with this possibility are in vitro studies in which TLR2 and TLR4 do not play a role in macrophage interactions with Histoplasma yeasts (Lin et al. 2010). Similarly, TLR2 does not contribute to the interferon (IFN)-β response of macrophages to Histoplasma conidia (Inglis et al. 2010). On the other hand, biochemical fractions of Histoplasma yeast cell walls can stimulate lipid body formation by macrophages (which is correlated with leukotriene production), and this is dependent on TLR2, indicating that cell wall fractions contain PAMP(s) recognized by TLR2 (Sorgi et al. 2009). Together, these data suggest that Histoplasma yeast have TLR ligands but that they are hidden from immune cells in intact, wild-type yeasts in much the same way that cell wall β-glucans are veiled (Fig. 7.1). All of these studies used cultured phagocytes so the role of TLRs in vivo remains undetermined. It is unknown whether α-glucan might also be the cell wall factor that masks the TLR ligands.
III. Negation of Host Defenses
A. Elimination of Antimicrobial Reactive Oxygen
Interaction of Histoplasma yeasts with phagocytes variably induces production of reactive oxygen species (ROS), which Histoplasma must detoxify in order to survive. Stimulatory signals, including PRR recognition of PAMPs, cytokines, and other phagocytic signals, trigger assembly of the NADPH-phagocyte oxidase (phox), a multimeric protein complex that produces the initial ROS, superoxide (O2 −). In response to Histoplasma yeasts, PMNs readily produce ROS (Schaffner et al. 1986; Schnur and Newman 1990; Kurita et al. 1991a, b) but macrophages only produce significant ROS upon cytokine activation (Eissenberg and Goldman 1987; Wolf et al. 1987, 1989; Fleischmann et al. 1990; Wolf and Massof 1990). Regardless, Histoplasma survives phagocyte-derived superoxide in vitro and in vivo by expression of a yeast-phase-specific, extracellular superoxide dismutase, Sod3 (Youseff et al. 2012). Histoplasma Sod3 is both secreted and anchored on the yeast cell wall, positioning it optimally to destroy phagocyte-derived superoxide (Fig. 7.2). Intracellular superoxide dismutase (i.e., Histoplasma Sod1) does not provide protection against extracellular superoxide because the charge on superoxide renders this ROS unable to pass through biological membranes. Without Sod3, Histoplasma virulence is significantly attenuated and Histoplasma yeast survival against PMNs and activated macrophages is reduced to similar levels as for wild-type Candida. Thus, Sod3-dependent dismutation of host-derived superoxide contributes to the greater virulence of Histoplasma compared to opportunistic fungi.
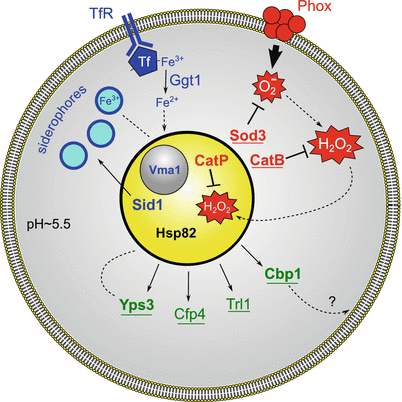
Fig. 7.2.
Mechanisms facilitating Histoplasma yeast intracellular survival and proliferation within macrophage phagosomes. Demonstrated virulence factors are indicated in bold and those with pathogenic-phase-specific expression are underlined. Histoplasma yeasts extinguish phagocyte-produced ROS by production of extracellular oxidative stress defense factors (red). Assembly of the multimeric phagocyte NADPH-oxidase complex (Phox) in the phagosomal membrane leads to production of antimicrobial superoxide. This toxic ROS is eliminated by the extracellular Histoplasma superoxide dismutase (Sod3). Peroxide molecules are similarly destroyed by extracellular catalase (CatB). Residual peroxide that escapes CatB destruction and enters the yeast cell is eliminated by an intracellular catalase (CatP). Histoplasma yeasts have at least two strategies for essential iron acquisition (blue). Host iron is brought to the phagosome via transferrin after binding to the transferrin receptor (TfR). In one strategy, Histoplasma yeasts secrete a γ-glutamyl transferase (Ggt1), which utilizes GSH to reduce ferric to ferrous iron, promoting iron release from transferrin and allowing for import into yeast cells. In a second strategy, the Histoplasma ornithine monooxygenase (Sid1) initiates siderophore synthesis and siderophores are secreted into the phagosomal lumen to chelate iron. The action of the vacuolar proton-ATPase, of which Vma1 is a central subunit, maintains iron homeostasis in Histoplasma yeasts. Histoplasma yeasts also secrete several novel factors (green) with undefined roles in intracellular residence. Cbp1, a calcium-binding protein, is abundantly secreted by yeast cells and may interact with phagosomal lipids. Yps3 is a secreted factor that also binds to cell wall chitin. Histoplasma yeasts produce an extracellular thioredoxin reductase-like protein (Trl1) and the Cfp4 glycoprotein during infection of macrophages. Additionally, yeasts utilize the stress-response Hsp82 protein to ameliorate intracellular stresses
In addition to Sod3, Histoplasma yeasts express a yeast-phase-specific, extracellular catalase, CatB. CatB is equivalent to M-antigen, a major diagnostic exoantigen for histoplasmosis (Hamilton et al. 1990; Zancopé-Oliveira et al. 1999). Given the ability of CatB to destroy peroxide, many have assumed that CatB is essential to Histoplasma virulence. However, CatB-deficient Histoplasma yeasts are fully virulent (Holbrook et al. 2013). This is due to functional redundancy with the intracellular Histoplasma catalase, CatP (Holbrook et al. 2013). Production of either catalase is sufficient to protect Histoplasma from host-produced ROS. Nonetheless, expression of dual catalases may provide a more effective means of protecting ROS-sensitive intracellular targets (Fig. 7.2); the phagocyte-derived ROS is efficiently destroyed by extracellular CatB before peroxide can enter the cell and affect intracellular targets. CatP, as an intracellular ROS defense, provides additional protection against any peroxide that escapes destruction by CatB. The combined activities of Sod3 and the dual catalase system effectively negate phagocyte-produced reactive oxygen defenses, thereby enabling yeast survival during infection.
B. Resistance to Reactive Nitrogen Compounds
Host defenses against fungal infections also include production of reactive nitrogen species (RNS), particularly by cytokine-activated phagocytes. Depletion or inhibition of the pro-inflammatory cytokines IFN-γ, TNF-α, and GM-CSF increases susceptibility to Histoplasma infection. This effect is correlated with impaired production of reactive nitrogen compounds, implicating RNS as one of the host defense mechanisms Histoplasma must combat (Lane et al. 1994; Allendoerfer and Deepe 1998; Deepe et al. 1999). Consistent with this, inhibition of RNS production by phagocytes in culture decreases their ability to control Histoplasma yeasts (Nakamura et al. 1994; Lane et al. 1994). However, the effect of phagocyte RNS on Histoplasma yeast is fungistatic, not fungicidal, suggesting that Histoplasma has defenses against the lethal effects of RNS. As candidate factors mediating this, 59 genes are upregulated in Histoplasma yeasts specifically in response to RNS treatment (Nittler et al. 2005). One of the induced genes is a mycelial-expressed nitric oxide reductase (Nor1) that, when ectopically expressed in yeasts, provides a slight benefit in vitro to yeast grown in the presence of nitric oxide. Whether Nor1 is necessary for the RNS resistance of Histoplasma yeasts remains to be determined.
C. Stress Response
In addition to ROS- and RNS-based host defenses, Histoplasma yeasts must contend with other stresses related to intracellular growth and host infection. A heat-shock protein (Hsp82), which is homologous to proteins of the 90 kDa stress-response family, facilitates intracellular growth as well as full virulence in vivo (Edwards et al. 2011b). Although it is assumed that heat-shock proteins are essential for thermally dimorphic fungal adaptations, yeasts with reduced Hsp82 function grow normally at mammalian body temperature (37 °C). At febrile temperatures, hsp82-mutant yeasts are more inhibited than wild-type yeast cells. Thus, Hsp82 is not required for yeast growth at 37 °C but instead ameliorates stresses associated with infection that are not limited to high temperature growth.
IV. Essential Nutrients for Intracellular Growth
A. Iron Acquisition
Like all cells, Histoplasma yeasts require iron for essential life processes and, during infection, must acquire this element from the host. Host iron restriction, enhanced by IFN-γ that is produced during cell-mediated immunity or administration of iron chelators, restricts Histoplasma replication (Lane et al. 1991; Newman et al. 1994, 1995). To obtain limited iron, Histoplasma relies upon a number of iron acquisition strategies. Maintenance of the phagosomal pH at around 5.5 is thought to allow release of one of the two iron atoms from transferrin, yet this pH is not acidic enough for activation of lysosomal hydrolases. In addition, Histoplasma yeasts produce and secrete iron-scavenging hydroxamate siderophores, which can acquire iron from holotransferrin (Howard et al. 2000) (Fig. 7.2). Recent genetic studies have confirmed the importance of siderophore production in Histoplasma virulence; the loss of siderophore production reduces Histoplasma yeast growth under iron-limiting conditions in vitro, and impairs yeast replication in cultured macrophages by 50–75% (Hwang et al. 2008; Hilty et al. 2011).
Conflicting evidence exists for the requirement of siderophores in vivo. In one study, loss of siderophore production in the genetically tractable Panama strain does not impair infection and replication of Histoplasma in murine lungs until 15 days post infection (Hwang et al. 2008). On the other hand, depletion of siderophores in the NAm 2 strain caused a nearly 100-fold reduction in lung fungal burden at 7 days post infection (Hilty et al. 2011). This discrepancy may result from the operation of alternative iron acquisition mechanisms in some Histoplasma strains. In support of this, the genome of the Panama strain, but not the NAm 2 strain, contains FET3 and FTR1 genes that encode a high-affinity iron transport system (Hilty et al. 2011).
Although iron is an essential cofactor, excess iron has detrimental effects on cells, and consequently iron acquisition must be tightly regulated. Iron limitation induces both transcriptional (Hwang et al. 2008) and protein level changes (Winters et al. 2008) in Histoplasma yeast. A cluster of genes repressed by the iron-responsive GATA transcription factor, Sre1, encode enzymes responsible for siderophore synthesis in response to iron limitation (Hwang et al. 2008, 2012). The enzyme ornithine monooxygenase (encoded by the SID1 gene) that catalyzes the first committed step in siderophore biosynthesis is included in this iron-regulated gene cluster. Iron homeostasis in Histoplasma also depends on vacuolar pH because loss of the vacuolar ATPase subunit Vma1 or treatment with chemicals that block vacuolar acidification (e.g., bafilomycin) impair growth under iron-limiting conditions (Hilty et al. 2008).
Histoplasma yeasts preferentially utilize ferrous iron (Zarnowski et al. 2008b) and have multiple ferric ion reducing systems (Fig. 7.2). Each of these ferric reducing systems is extracellular, consistent with reduction of ferric ions to promote release from extracellular siderophores and host iron-carrying molecules such as transferrin. Early biochemical studies indicated that yeasts produce three extracellular ferric reductases, two soluble and one cell-associated (Timmerman and Woods 1999, 2001). The secreted glutathione-dependent ferric reductase has now been identified as a γ-glutamyl transferase (Ggt1), which is produced by Histoplasma yeast cells and catalyzes a two-step, pH-independent reduction of ferric iron (Zarnowski et al. 2008a) Depletion of Ggt1 by RNAi impairs the ability of Histoplasma yeasts to replicate and kill cultured phagocytes but the requirement for Ggt1 in vivo remains to be determined.
B. De Novo Vitamin Biosynthesis
Growth of Histoplasma yeasts in the nutrient-limiting phagosome of phagocytes requires de novo vitamin synthesis. Growth in chemically defined, minimal media confirms that Histoplasma yeasts can synthesize the essential cofactors riboflavin, niacin, pantothenate, pyridoxine, biotin, and folate (McVeigh and Morton 1965). On the other hand, mammals are unable to synthesize these vitamins and must acquire them from dietary sources. Yeast cells unable to synthesize their own riboflavin are severely compromised for intracellular growth (Garfoot et al. 2013). These yeasts are similarly unable to replicate following lung infection, indicating that dietary vitamins of the host are unavailable to intracellular yeasts in vivo, therefore mandating that Histoplasma yeasts synthesize them de novo. This suggests that targeting vitamin biosynthetic enzymes is a viable strategy for development of new therapeutics.
V. Secretion of Novel Factors
To influence the outcome of the host–pathogen interaction, Histoplasma yeasts secrete several novel factors with as yet unidentified functions (Fig. 7.2). Each of these factors is preferentially expressed by pathogenic-phase cells compared to nonpathogenic mycelia, arguing that their function is fundamentally linked to pathogenesis. The secreted Cbp1 protein was the first virulence factor identified and, without it, yeasts are significantly attenuated in their ability to establish respiratory infection (Sebghati et al. 2000). Originally described as a calcium-binding protein (Batanghari and Goldman 1997), Cbp1 has nanomolar affinity for calcium (Beck et al. 2008). The Cbp1 protein is a homodimer that is held in a compact structure by intramolecular disulfide bonds, making Cbp1 highly resistant to protease treatment, a characteristic that suits it well for function in the macrophage phagosome (Beck et al. 2008). Indeed, Cbp1 is secreted into the lumen of the phagosome by yeast cells (Kügler et al. 2000). The α-helices of Cbp1 have structural similarity to lipid-binding saposin-family proteins, which suggests that Cbp1 interacts with host membranes or glycolipids of the phagosome (Beck et al. 2009).
Histoplasma yeasts secrete the Yps3 protein, although production is restricted to the North American (NAm 2) phylogenetic group (Keath et al. 1989; Keath and Abidi 1994; Bohse and Woods 2007a). Restricted production results from transcriptional regulation because replacement of the promoter of the YPS3 gene with a constitutive promoter restores production of Yps3 in Histoplasma cells that normally do not synthesize it (Bohse and Woods 2007a). Yps3 has homology to the Bad1 Blastomyces adhesin protein and, like Bad1, Yps3 is both secreted and associated with the cell wall through interactions with chitin (Weaver et al. 1996; Bohse and Woods 2005). However, Yps3 does not share the adhesin functions of Bad1 and, although Yps3 has internal tandem repeats, Yps3 lacks the tryptophan-enriched tandem repeat domains of Bad1 that have immunomodulatory function. Functionally, Yps3 is dispensable for pulmonary colonization, but depletion of Yps3 by RNAi impairs the ability of yeasts to establish disseminated infections (Bohse and Woods 2007b). At the cellular level, no function has yet been identified for either soluble or cell-associated Yps3.
A more systematic effort to identify the proteins comprising the secreted proteome of Histoplasma yeasts has revealed additional factors with potential roles in Histoplasma virulence (Holbrook et al. 2011). The secreted proteome includes the Sod3, CatB, and Cbp1 factors described above. Additional prominent factors in the extracellular fraction include glycanases (exo- and endoglucanases, Exg1 and Eng1, respectively) as well as a chitinase (Chs1), which probably contribute to yeast cell wall remodeling. The secreted proteome has at least five novel culture filtrate proteins (Cfp). Chief among these is Cfp4, a copiously produced, heavily glycosylated factor uniquely expressed by yeast-phase cells. Histoplasma yeasts also secrete a thioredoxin reductase-like protein (Trl1), which is distinct from intracellular thioredoxin reductases. Although no functional role has been determined for the glycanases, Cfp4, or Trl1, expression of each factor is linked to the pathogenic phase and expression is maintained during infection (Holbrook et al. 2011). Given that these factors share characteristics common to the vast majority of demonstrated virulence factors (i.e., extracellular localization and pathogenic-phase regulated expression), the identified secreted proteome is a rich source of candidate factors that promote Histoplasma virulence. In addition to the canonical eukaryotic secretion pathway, Histoplasma also secretes vesicles (Albuquerque et al. 2008), which may be a secondary means of delivering factors to the extracellular environment. Nevertheless, not all secreted factors are necessarily important for virulence, as shown by the lack of any attenuation when the secreted dipeptidyl peptidase (DppIV) is depleted (Cooper and Woods 2009).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


