Molecular and Cellular Biology of the Normal, Hypertrophied, and Failing Heart: Introduction
The human heart has a tremendous capacity to change its size and shape in response to external stimuli.1 During embryonic development and the postnatal state, the heart grows via hyperplasia (increase in cell number) and hypertrophy (increase in individual cell size). In the adult heart the predominant form of growth is via cellular hypertrophy, although recent data suggest that cardiomyocyte replication and replenishment is possible in the adult myocardium.2,3 Growth signals that occur in the setting of postnatal maturation, pregnancy, and endurance exercise can lead to physiologic hypertrophy, a process where the heart grows with preservation of overall structure and function. In contrast, stimuli such as mechanical overload, ischemia, diabetes, and sarcomeric protein mutations can lead to pathologic hypertrophy, a process where growth is associated with abnormalities in cardiac geometry, performance, tissue architecture, and cellular function. From a clinical standpoint, physiologic hypertrophy has no adverse sequelae, whereas pathologic hypertrophy is associated with increased risk of heart failure, arrhythmias, and death. In addition to its capacity for growth, the heart can shrink its mass in response to mechanical unloading or physical inactivity in a process termed cardiac atrophy. These phenotypic profiles have a robust dynamic range that approaches 100% and thus highlight the remarkable plasticity of the adult heart (Fig. 7-1).4 The application of contemporary biologic approaches is progressively elucidating the cellular and molecular pathways that drive cardiac hypertrophy, atrophy, and altered cardiac function.1 Precise definition of these pathways forms the foundation for novel heart-failure therapies.
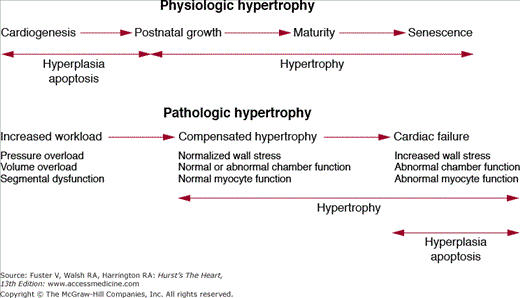
Cardiac Growth and Physiologic versus Pathologic Hypertrophy
Cardiac growth during normal development (also referred to as cardiac eutrophy) includes fetal cardiogenesis, postnatal cardiac growth, and the modest additional increase in heart size that evolves during senescence. The earliest stage of cardiac growth in utero is governed by a genetically determined developmental program which can occur in the absence of contractile activity. Subsequent fashioning of the developing heart is determined by an intricate interplay between genetic programs and mechanical forces. The fetal four-chambered mammalian heart attains an adult structural configuration in the second trimester, but continues to enlarge to maintain circulatory support for the growing embryo and juvenile.5 In rodents and other experimental models, fetal myocardial growth is largely a consequence of increasing number of cardiomyocytes (hyperplasia) until shortly after birth, after which cell division gradually subsides and cardiac mass increases almost entirely through enlargement of cardiomyocytes (hypertrophy).6 When hyperplasia subsides, many cells undergo a final round of karyogenesis (nuclear division) without cytokinesis (cell division) thereby producing a mixture of mononucleate and binucleate cardiomyocytes. Growth of the left ventricle exceeds that of the right ventricle during the early postnatal period as the mammalian heart transitions from the fetal- to adult-type circulation. Thereafter, the heart undergoes a six-fold increase in mass. The heart-to-body weight ratio then remains relatively constant throughout adolescence and adulthood.
In the adult heart, cardiomyocytes make up one-third of the cell number but are responsible for more than 70% of cardiac volume. In the adult, increases in cardiac mass are produced largely by an increase in size of fully differentiated cardiomyocytes. Adult cardiomyocytes have long been viewed as terminally differentiated—that is, incapable of reentering the cell cycle—and there is little evidence that they are capable of cell division under normal conditions after the early postnatal period. However, there is emerging evidence suggesting that a small subpopulation of cardiomyocytes can reenter the cell cycle and proliferate,3,7 although the clinical significance of this finding is currently uncertain. One recent study has demonstrated that the growth factor neuregulin-1 can induce mononucleate adult cardiomyocytes to proliferate in a mouse and thereby promote myocardial regeneration after infarction.2 The capacity to reactivate hyperplasia in the differentiated cardiomyocyte is an area of intense research interest with important therapeutic implications in the hypertrophied and failing heart.3
In response to stimuli such as chronic endurance exercise or pregnancy, the heart hypertrophies without alterations in structure and function. Indeed, the association of intense athletic conditioning with cardiac hypertrophy has been recognized for more than a century.8 Physiologic hypertrophy of the heart is not associated with heart failure or other adverse clinical sequelae.9 Morphologically, it is characterized by mild augmentation in wall thickness and cavity size and is associated with increase in the length-to-width ratio of cardiomyocytes. This form of cardiac growth is not associated with altered excitation-contraction coupling, metabolic dysregulation, cardiac fibrosis, or arrhythmia susceptibility. In humans, physiologic hypertrophy has been best studied in high-performance endurance athletes.10 Of all athletes, cross-country skiers, rowers, and cyclists tend to have the largest hearts, although most of them have a left ventricular wall thickness of less than 1.3 cm (upper limit of normal, 1.1 cm in adults who are not highly conditioned).4,10 These increases in wall thickness are associated with mild left ventricular cavity dilation with a 10% to 15% augmentation in end-diastolic dimension. Even in the most conditioned athletes, LV wall thickness of greater than 1.3 cm without some degree of associated cavity dilation is unusual and should raise suspicion for a primary pathologic state such as hypertrophic cardiomyopathy.10 Epidemiologic data fail to demonstrate adverse risk associated with the modest hypertrophy that occurs as a consequence of athletic conditioning. Thus it is important clinically to distinguish physiologic hypertrophy resulting from physical training from other forms of cardiomyopathy. Physiologic hypertrophy can regress to baseline levels after cessation of intense training11 or in the postpartum period.12
Many clinically relevant pathologic stimuli, including hypertension, coronary insufficiency, or valvular malfunction, first induce a phase of compensated cardiac hypertrophy in which individual myocytes grow in length and/or width as a means to maintain cardiac pump function and ameliorate ventricular wall stress.13 However, these initially adaptive responses are associated with significant perturbations in myocardial gene expression, architecture, and physiology. In both humans and animal models, ventricular cardiomyocytes undergoing pathologic hypertrophy reinduce a subset of genes that are normally expressed at high levels during fetal life. This “fetal gene program” includes genes such as atrial natriuretic peptide, alpha-skeletal actin, and fetal isoforms of the myosin heavy chain.13 Cardiomyocytes undergoing pathologic hypertrophy also recapitulate the fetal metabolic program with decreased rates of fatty-acid oxidation and increased rates of glucose oxidation. Other pathologic changes at the cellular level include reorganization of the sarcomere, altered calcium homeostasis, changes in contractility and relaxation, death of cardiomyocytes with fibrotic replacement, and “electrical remodeling” (alterations in the expression or function of ion-transporting proteins).4
An established geometric classification scheme describes cardiac hypertrophy as occurring in a concentric or eccentric pattern. Concentric hypertrophy is typically triggered by pressure overload (eg, chronic hypertension), which results in significantly elevated systolic wall stress. At the organ level, concentric hypertrophy is characterized by an increase in relative wall thickness and cardiac mass with little or no change in chamber volume. At the cellular level there is increased cardiomyocyte cross-sectional area with addition of sarcomeres in parallel, thereby causing predominantly lateral growth of individual cells. Eccentric hypertrophy is typically triggered by volume overload (as occurs in chronic valvular regurgitation), which results in increased diastolic and systolic wall stress. Regional hypertrophy that develops in viable myocardium after a myocardial infarction also occurs in an eccentric pattern. At the organ level, eccentric hypertrophy is characterized by an increase in cardiac mass with increased chamber volume. Relative wall thickness may be normal, decreased, or increased. At the cellular level, there is addition of sarcomeres in series with predominantly longitudinal myocyte growth. The precise cellular mechanisms responsible for concentric versus eccentric growth patterns are not well understood. Although this classification scheme helps to characterize the stereotypic response of the heart to a given stimulus, many diseases produce overlapping patterns of hypertrophy. For example, in chronic hypertension or aortic stenosis, the heart can initially develop concentric hypertrophy that transitions to eccentric hypertrophy in advanced stages. However, successive progression through these stages is not always the case as illustrated by postinfarction cardiomyopathy or familial dilated cardiomyopathies, which can directly progress to severe eccentric hypertrophy without a concentric phase. Regardless of growth pattern, if the stimulus for hypertrophy is sufficiently intense or prolonged, decompensated hypertrophy and clinical heart failure can ensue.
Whereas cardiac hypertrophy is commonly triggered by changes in ventricular loading, there are instances where distinct molecular perturbations within the cardiomyocyte are sufficient to initiate the hypertrophic response in the face of normal loading conditions. In some forms of familial hypertrophic cardiomyopathy that result from point mutations in sarcomeric proteins, the ventricle appears to hypertrophy independent of external wall stress.14 As a second example, mice genetically engineered to express excessive amounts of the protein phosphatase calcineurin develop severe cardiac hypertrophy and heart failure despite unchanged extracardiac loading conditions.15 Experimental studies in the past decade have called into question the idea that the initial phase of cardiac hypertrophy is adaptive and protective.4,16 New ways of interrupting myocardial signaling in mice have made it possible to curb or abolish hypertrophic growth without altering the inciting growth-stimulus pathways. Attenuation or elimination of hypertrophy in the face of pressure-overload is often surprisingly well tolerated in these animal models—ventricular dilation or decompensation does not develop.17 The studies raise the possibility that modulating cardiac growth pathways can provide clinical benefit without significant hemodynamic deterioration.4,17 These and other disparate observations suggest a critical reexamination of the primary role of mechanical load in the etiology of certain forms of pathologic hypertrophy.
Cardiac Atrophy
The heart can decrease its net mass (cardiac atrophy) in the setting of mechanical unloading, disuse, and catabolic endocrine signals (eg, hypothyroidism and hypopituitarism).18 After a chronically stenotic aortic valve has been replaced, the hypertrophied human left ventricle can regress its mass within several weeks postoperatively.19 In a canine model of ventricular unloading induced by contraction of the inferior vena cava, a 26% decrease in left ventricular mass occurs in 10 days.20 In the clinical setting, cardiac atrophy can also occur with prolonged bed rest,21 extreme physical inactivity,22 microgravity conditions,23 and the use of mechanical assist devices for advanced heart failure.24 An important animal model used to study unloading-induced cardiac atrophy is the heterotopically transplanted heart. This model can be applied to the normal, ischemic, or hypertrophied heart and resembles in many ways the failing human heart supported by a mechanical-assist-device.18,25,26
Cardiac atrophy is an energy-requiring process in which progrowth pathways are suppressed27 and protein-degradatory pathways are induced.28 The molecular underpinnings of cardiac atrophy are just beginning to be deciphered and much of our knowledge is derived from the study of skeletal muscle atrophy.4,18 A common molecular mechanism for muscle atrophy is the activation of the ubiquitin-proteasome system, an energy-dependent cellular pathway that regulates the selective and efficient degradation of proteins.29 The atrophic process requires the activation of ubiquitin ligases (also known as atrogenes), a large family of enzymes that selectively target individual proteins for cellular degradation.30 Study of these degradatory genes has provided important insights into cardiac atrophy as well as the day-to-day protein-quality control that is necessary for cardiomyocyte homeostasis.29 A better understanding of these antigrowth and proatrophic pathways may lead to novel therapies for pathologic hypertrophy and heart failure.
Model Systems
A significant portion of this chapter discusses the molecular pathways that regulate cardiac hypertrophy. While critical insights can be gained from studies in cultured cells, the advent of genetic engineering in the mouse has ushered in a new era of investigative power in heart disease.31 Cardiac hypertrophy and heart failure are whole-organ phenomena that are in constant interplay with circulating factors and other organ systems. As such, genetically modified mouse models, if designed and employed properly, can elucidate disease-gene relationships and define novel therapeutic targets.31 In this regard, the mouse has emerged as the premier mammalian model system because its genetics and development have been well characterized, it allows precise manipulation of its genome, and has a short gestation period.32,33 More detail about the exact nature of these techniques is provided in Chap. 6.32 This chapter briefly summarizes the broad approaches used to probe gene function in the heart of an intact organism.
An investigator trying to understand the role of a single gene in a process such as cardiac hypertrophy asks two critical questions: (1) Is the gene necessary for the development of cardiac hypertrophy? (2) Is increased function of the gene sufficient to drive cardiac hypertrophy? The question of necessity is addressed using loss-of-function experiments in which a gene can be inhibited pharmacologically or ablated genetically (gene targeting or “knockout” technology). The question of sufficiency is addressed using gain-of-function approaches in which a gene can be activated by a drug or forcibly overexpressed in the heart using genetic engineering (transgenesis). Contemporary genetic engineering technology has allowed investigators to ablate or overexpress genes only in a particular cell type or at a particular time of interest. These methods can help dissect a gene’s spatiotemporal effects with increased precision.32 In addition, certain disease-causing human gene mutations can be introduced into the homologous location in the mouse genome to create a human disease model. Such an approach for modeling monogenic human disease has been successfully applied to familial hypertrophic cardiomyopathy34 and has led to significant clinical advances.
There are also a number of tools that scientists use to characterize experimental animals in an attempt to model human disease.32 It is possible to induce pressure-overload cardiac hypertrophy in rodents using surgical aortic constriction. Agents such as angiotensin II (ANG-II), norepinephrine, endothelin 1 (ET-1), thyroxine, or insulin-like growth factor I (IGF-I) can also be chronically infused into animals to model neurohormone-induced hypertrophy. Coronary ligation can be performed to simulate postinfarct hypertrophic remodeling. Animals can be subject to chronic endurance exercise to simulate physiologic hypertrophy. Initially, the small size of the murine heart was a challenging and limiting factor in the physiological and pathological evaluation of phenotypic cardiac alterations. However, advances in “microphysiology” have provided opportunities to assess cardiac function in an integrative approach from the cellular to the intact animal level.32,35 In addition to standard molecular and histologic methods, these phenotyping tools include two-dimensional echocardiography, cardiac MRI, invasive hemodynamic assessment with pressure-volume analysis, metabolic exercise studies, continuous electrocardiographic telemetry, and electrophysiological studies. Taken together, the ability to manipulate the mouse genome and precisely characterize physiologic responses has been instrumental in defining the molecular effectors of cardiac hypertrophy and heart failure (Fig. 7–2).
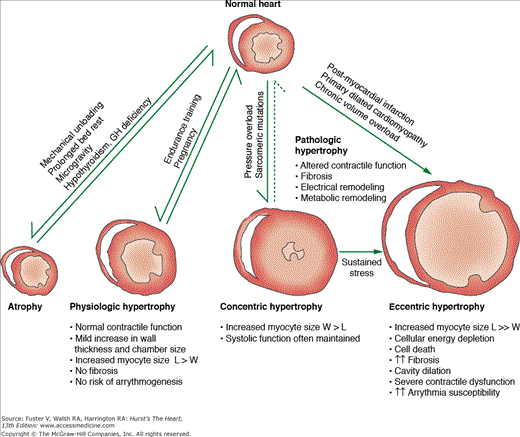
Molecular Mechanisms of Cardiac Hypertrophy
While the molecular pathways that drive cardiac hypertrophy are woven into complex networks, signaling passes through a limited number of common nodal points’some at the cell surface, some in the cytoplasm, and some in the nucleus.1,4 This section provides an overview of these signaling mechanisms and highlights these nodal points. At the cell surface, biomechanical forces and neurohormones stimulate receptors to generate the most proximal signals. These receptors then relay biochemical messages to the cell interior using a variety of enzymatic cascades in a process termed signal transduction. Important classes of these downstream messengers are kinases, proteins that can add phosphate groups to target macromolecules and modulate their function. Many of these signaling pathways can alter the biology of the cardiomyocyte acutely by affecting proteins involved in calcium homeostasis, excitation–contraction coupling, energy utilization, redox balance, and cell survival. However, these signals also converge on the cell nucleus where they are integrated to produce alterations in gene expression by a group of proteins termed transcriptional regulators. These transcriptional regulators have access to the cell’s genome and function as genetic switches that can turn genes on and off in a signal-dependent manner. The repertoire of expressed genes (the transcriptome) ultimately determines the characteristics of the cell. A major focus of current research is to identify nodal points at all three cellular levels with an eye toward therapeutic manipulation (Fig. 7-3).
Figure 7–3
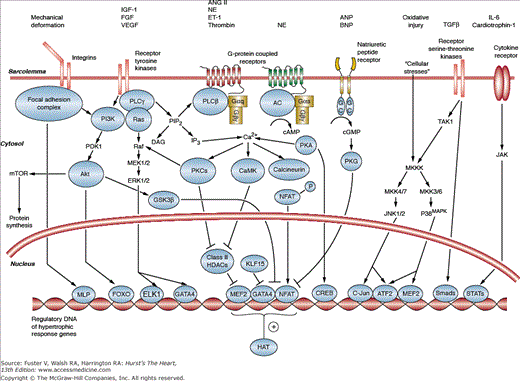
A schematic outlining some of the major overlapping signal transduction pathways that mediate cardiac hypertrophy. There are important signal transducers at all three cellular locations: the sarcolemmal membrane, the cytosol, and the nucleus. Diverse signals ultimately converge upon the nucleus where they are integrated by gene-regulatory proteins to produce long-term alterations in cardiac structure and function. Details of individual pathways are provided in the text. For sake of clarity, the full extent of crosstalk between these pathways has been omitted. IGF-I, insulin-like growth factor I; FGF, fibroblast growth factor; VEGF, vascular endothelial growth factor; Ang-II, angiotensin II; NE, norepinephrine; ET-1, endothelin 1; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; IL-6, interleukin-6; PI3K, phosphoinositide 3-kinase; PLC, phospholipase C; AC, adenylate cyclase; cAMP, cyclic adenosine monophosphate; GC, guanylate cyclase; cGMP, cyclic guanosine monophosphate; PKG, cGMP dependent protein kinase or protein kinase G; TGF-b, transforming growth factor beta; TAK1, transforming growth factor b–associated kinase-1; JAK, Janus kinase; mTOR, mammalian target of rapamycin; PDK1, phosphoinositide dependent kinase 1; GSK3b, glycogen synthase kinase 3b; DAG, diacylglycerol; IP3, inositol triphosphate; PKA, cAMP dependent protein kinase or protein kinase A; MAPK, mitogen-activated protein kinase; JNK, c-jun N-terminal kinase; CaMK, calcium-calmodulin dependent protein kinase; NFAT, nuclear factor of activated T-cells; MEK1/2, mitogen-activated protein kinase-1/2. ERK1/2, extracellular signal-regulated kinase-1/2; MLP, muscle LIM protein; FOXO, forkhead box transcription factor class O; ELK1, Ets like gene-1; MEF2, myocyte enhancer factor-2; CREB, cAMP response element binding protein; ATF2, activating transcription factor-2 or activating protein-2; STAT, signal transducers and activators of transcription; HAT, histone acetyltransferase; HDAC, histone deacetylase.
Mechanical loading of cultured cardiomyocytes (eg, cyclic deformation) is sufficient to cause cellular hypertrophy.36 The process by which cells sense these mechanical stimuli and convert them into biochemical signals is termed mechanotransduction. Cardiomyocyte mechanotransduction appears critically important for the development of both physiologic and pathologic hypertrophy. The cardiomyocyte detects mechanical stimuli via a number of proximal molecular sensors including sarcomeric protein complexes, the cytoskeleton, stretch activated ion channels, ligand-activated cell surface receptors, and membrane proteins involved in interactions with the extracellular matrix (ECM). Recent evidence points to the integrin family of proteins as one important component of the cardiomyocyte mechanosensing apparatus. Integrins are a large family of heterodimeric transmembrane receptors that bind to the extracellular matrix and link to the intracellular cytoskeleton.37 In the setting of mechanical activation, integrins can cluster on the cell surface and directly stimulate proximal effectors such as focal adhesion kinase (FAK), the small GTPase Rho, and the adaptor protein melusin. These proximal signals activate a cascade of downstream pathways that result in enhanced protein synthesis, myofibrillar assembly, hypertrophic gene transcription, cell survival, and angiogenesis. In addition, integrin stimulation can signal the myocyte to produce and release peptide growth factors such as ANG-II, VEGF, and ET-1 that can act on their cognate receptors in an autocrine/paracrine fashion. This autocrine/paracrine mechanism may indeed be responsible for a considerable portion of the growth response of cultured cardiomyocytes to mechanical stretch in vitro and the intact heart to pressure overload in vivo.38 A second mechanosensing apparatus has been proposed at the level of the Z-disc within each sarcomere. Specifically, the small LIM-domain protein MLP (muscle LIM protein), which is anchored to the Z-disc, is thought to function as an internal stretch sensor.13,39 The genetic deletion of each of these proximal deformation-sensor proteins (integrin, FAK, melusin, and MLP) in mice leads to cardiac dilation and dysfunction, showing the importance of these molecules in the preservation of cardiac homeostasis and the compensatory response to stress.13,40-44
Excessive neurohormonal stimulation with factors such as ANG-II, norepinephrine, and ET-1 is now widely accepted as a central feature of pathologic hypertrophy and heart failure in humans. These neurohormones bind to specific seven-transmembrane-spanning receptors that couple to heterotrimeric G-proteins of the Gαq/α11 subclass. Gαq is coupled to an enzyme termed phospholipase-Cβ (PLCβ), which cleaves a cellular phospholipid into two highly active lipid signaling molecules: diacylglycerol (DAG) and inositol(1,4,5) triphosphate [Ins(1,4,5)P3].45 DAG binds and activates a family of critical intracellular kinases of the protein kinase C family (PKC) which can signal to the nucleus to promote hypertrophic gene expression.46 Ins(1,4,5)P3 can bind to a specific receptor on the sarcolemmal or nuclear membrane and trigger release of intracellular Ca2+ stores. These net increases in intracellular calcium can activate a myriad of Ca2+-dependent hypertrophic pathways including the nuclear factor of activated T-cells (calcineurin-NFAT) circuit and calmodulin-dependent kinases (CaMKs), both of which can directly signal to the nucleus. Studies in genetically modified mouse models have confirmed that Gαq/α11 coupling is both necessary and sufficient for the development of pathologic hypertrophy. Forced overexpression of Gαq in the mouse heart can drive pathologic hypertrophy and cause heart failure.47-49 Conversely, the combined disruption of Gαq and Gα11 signaling50 or overexpression of a Gαq-inhibiting protein51 both blunt the hypertrophic response to pressure overload with maintenance of LV systolic function. Importantly, these studies indicate that Gαq-dependent hypertrophy is not required for functional compensation to stress and support the targeting of G protein–coupled receptor (GPCR)-Gαq signaling as a therapeutic strategy.
Beta adrenergic receptors (β-ARs) are seven transmembrane receptors that couple to heterotrimeric proteins of the Gα family. Dysregulation of β-AR signaling is a universal feature of human heart failure and pharmacologic inhibition of these receptors is a mainstay of therapy.52 β1-ARs couple to Gαs proteins and activate adenylate cyclase to produce a rise in intracellular cyclic adenosine monophosphate (cAMP). cAMP activates a particular cytosolic kinase termed cyclic-AMP dependent protein kinase (PKA) whose downstream targets include L-type calcium channels, phospholamban, and the ryanodine receptor. The net effect of PKA stimulation is to dramatically increase intracellular calcium levels thereby inducing inotropic and chronotropic responses as well as activating a broad range of calcium-sensitive signaling molecules that can induce pro-hypertrophic gene expression.
In addition to the canonical pathways described above, it appears that certain GPCRs can signal through important alternative mechanisms. For example, in the setting of biomechanical stress, the ANG-II type 1 receptor (AT1R) can directly associate with Janus kinase-2 (JAK2) in a ligand-independent manner and induce translocation of Gαq into the cytosol.53 In addition, the AT1R can signal to the mitogen-activated protein kinases (MAPKs) independent of Gαq coupling via mechanisms that are not completely understood. This cross talk between the AT1R and MAPK signaling appears to be an important stimulus for pathologic hypertrophy.54,55 Research over the last 2 decades has defined a critical role for the beta-gamma (βγ) subunits of the heterotrimeric G-proteins in a myriad of biologic processes.56 In the cardiomyocyte, βγ-dependent signaling can activate members of the phosphatidyl-inositol 3-kinase (P13K) family of lipid kinases thereby providing crosstalk between the adrenergic and growth factor (eg, insulin, VEGF) signaling pathways. Importantly, both Gα- and βγ-dependent signaling initiate a complex series of intracellular events whose net result is to desensitize and internalize the β1-AR.57 Given the observation that the β1-AR is desensitized in human heart failure57 and the consistent clinical efficacy of β1-AR antagonists in the treatment of heart failure,52 a better understanding of these noncanonical signaling pathways has significant therapeutic implications (Fig. 7–4).
Figure 7–4
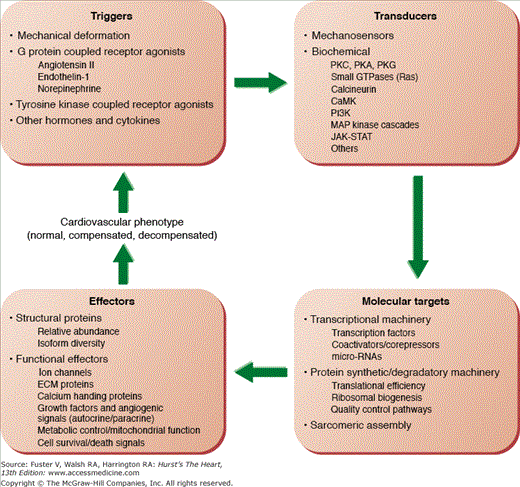
Schematic diagram of the mechanisms responsible for the development of the anatomical and functional cardiac phenotypes in physiologic and pathologic hypertrophy. Abnormalities in one or multiple levels in this putative closed-loop system may be responsible for the transition between compensated and decompensated hypertrophy.
Growth factors such as insulin, (IGF-I), fibroblast growth factor (FGF), and vascular endothelial growth factor (VEGF) bind cell surface receptors that have intrinsic tyrosine kinase or serine/threonine kinase activity in their cytoplasmic domains. In the cardiomyocyte, these receptors are coupled to three major signal transducers: PI3K, phospholipase C, and the Ras family of small GTPases. Within this family, the IGF-1/PI3K signaling-pathway and its role in physiologic hypertrophy has been best studied. There is substantial evidence based on studies performed in cultured cardiomyocytes and genetically-manipulated mice that IGF-1/PI3K signaling plays an important role in physiologic hypertrophy of the heart during normal development and in response to exercise training.58 Cardiac-specific overexpression of IGF-1 receptors in the mouse heart produces physiologic hypertrophy indistinguishable from that produced by exercise training.59 Ligand binding to the IGF-1 receptor activates PI3K of the subtype IA (PI3Kα).60 PI3K converts the plasma membrane lipid phosphatidylinositol-4,5-bisphosphate [PtdIns(4,5)P2] to phosphatidylinositol-3,4,5-trisphosphate [PtdIns(3,4,5)P2], which activates other signaling constituents in the plasma membrane.60,61 PI3Kα is a heterodimer that consists of a p85 regulatory subunit and a p110 (α, α, or δ) catalytic subunit.60 Forced expression of a constitutively active form of PI3Kα in the mouse heart is sufficient to produce a form of physiologic hypertrophy with preserved LV function.62 Conversely, myocardial expression of a dominant-negative p100a transgene results in blunted physiologic hypertrophy associated with postnatal growth and exercise, while the hypertrophic response to pressure-overload is unchanged.58 Similarly, mice with germline deficiency of the p85 regulatory subunit have smaller hearts at baseline and after exercise.63 Despite the lack of effect on cardiac mass, the loss of P13Kα function in the heart leads to cardiac dilation and ventricular dysfunction with pressure overload, suggesting that PI3Kα exerts protective effects in the face of stress.58
A critical downstream target of PI3Kα-signaling in the heart is the protein kinase B or AKT kinase (PKB/AKT). PI3K activation results in the sarcolemmal recruitment of phosphoinositide-dependent kinase (PDK1) and AKT and subsequent phosphorylation and activation of AKT by PDK1.61 Genetic deficiency of the Akt1 gene in mice results in attenuated physiologic hypertrophy with exercise and cardiac decompensation with pressure-overload.64 This phenotype resembles that seen with p110α loss-of-function and strongly suggests that Akt1 is a major effector of PI3Kα and physiologic hypertrophy in the heart.64 While studies of Akt1-transgenic mice suggest that this gene coordinates myocardial growth with coupled angiogenesis, prolonged AKT overexpression causes cardiac dysfunction.65 Therefore, it is likely that there is a critical dose and temporal window during which AKT activation exerts physiologically beneficial effects. There are two downstream targets of AKT that are important regulators of hypertrophic signaling: glycogen synthase kinase 3-beta (GSK3β) and the mammalian target of rapamycin (mTOR). In the cardiomyocyte, GSK3β is normally active under basal conditions and negatively regulates a number of key prohypertrophic transcription factors such as NFAT, GATA4, and c-Myc. In fact, overexpression of an active form of GSK3β in the mouse heart can attenuate hypertrophy in response to both mechanical and neurohormonal stimuli.66,67 AKT-mediated phosphorylation inactivates GSK3β and thereby releases inhibition of these potent transcription factors. AKT can also enhance protein synthesis by phosphorylating and activating mTOR.61,68 mTOR induces protein synthesis by activating ribosomal biosynthesis and by improving ribosomal translation efficiency. The inhibition of mTOR by rapamycin has been shown to protect against pathologic cardiac hypertrophy,65,69,70 however the precise downstream targets that mediate its hypertrophic effects in vivo are not fully characterized.
Cytokines such as interleukin-6 (IL-6) and cardiotrophin have been implicated in cardiac hypertrophy by a number of in vitro and in vivo methods. IL-6 and cardiotrophin activate the cardiomyocyte glycoprotein-130 (gp130) transmembrane receptor and rapidly stimulate cytoplasmic Janus kinases (JAKs); these, in turn phosphorylate other cytoplasmic proteins called STATs (signal transducers and activators of transcription). Various components of the gp130 and JAK-STAT pathway can induce cardiac hypertrophy when overexpressed in the mouse heart.71 By contrast, the cytokine TNFα (tumor necrosis factor alpha) signals via a distinct pathway that involves activation of the NFκB transcriptional pathway as well as of a phosphatidylcholine-specific isoform of phospholipase C that generates intracellular diacylglycerol. Despite the fact that a number of these cytokines are elevated in patients within the plasma of patients with congestive heart failure, a clinical trial using a TNFα inhibitor was halted due to lack of therapeutic benefit and increased adverse events.72
The MAP-kinases signaling system is comprised of a series of hierarchical kinases that transduce signals through successive phosphorylation. In rodents and humans, there are at least four tiers of kinases with each tier containing a number of family members. The most upstream kinases are called MAPKKKK (MAP kinase-kinase-kinase-kinases) and the most downstream kinases are the MAPKs. At its most proximal limb, the signaling cascade is initiated in cardiac myocytes via diverse stimuli including mechanical stretch, GPCRs, receptor tyrosine kinases, receptor serine/threonine kinases, and gp130 activation.13,73 These signals relay via a complex cascade of successive kinases that ultimately converge on three major distal MAPKs: ERKs (extracellular signal-regulated kinases), p38-MAPKs, and JNKs (c-Jun N-terminal kinases). Activation of ERKs, p38, and JNKs then phosphorylate diverse intracellular targets, including transcription factors that reprogram cardiac gene expression.13 Many of the kinases upstream of ERK, p38 and JNK form a complex network that has significant cross talk with other signaling modules. Thus mapping the precise signaling trajectories downstream of these proximal kinases has been challenging. The remainder of this section discusses the role of the most downstream kinases as signal integrators for this pathway.
Activation of ERK1/2 in the heart (using transgenic overexpression of an activated form of the ERK1/2-specific kinase, MEK1) leads to cardiac hypertrophy in vivo.74 Interestingly, this model produced concentric left ventricular hypertrophy with preserved systolic function and no evidence of cardiac fibrosis, suggesting that ERK1/2 activation results in compensated cardiac hypertrophy. Further mechanistic studies demonstrate that MEK1-ERK1/2 can crosstalk with the Calcineurin-NFAT circuit to coordinate the hypertrophic response in a codependent fashion.75 In contrast, activation of ERK5 (using transgenic overexpression of an activated form of its upstream MAPKK, MEK5) produces eccentric hypertrophy, dilated cardiomyopathy, and sudden death that is associated with the addition of sarcomeres in series within individual cardiomyocytes.76 These disparate phenotypes illustrate that activation of specific MAPKK-MAPK signaling modules can drive different forms of cardiac hypertrophy and underscores the importance of further mechanistic studies in the intact organism.
In contrast to the MEK1-ERK1/2 or MEK5-ERK5 pathways, activation of p38 or JNK signaling does not induce cardiac hypertrophy in mouse models. Transgenic expression of activated forms of MKK3 or MKK6, both of which specifically activate p38, lead to spontaneous postnatal heart failure with ventricular dysfunction, myocardial fibrosis, and wall thinning without net differences in cardiac mass.77 Similarly, transgenic mice that express activated MKK7 in the heart show specific activation of JNK and display cardiomyopathy without hypertrophic growth.78,79 Loss-of-function experiments that target p38-MAPK or JNK signaling lead to excessive pathologic hypertrophy, severe functional decompensation, and cardiomyocyte death.80-83 These effects might be driven, in part, by excessive activity of the Calcineurin-NFAT pathway.84 Taken together, these gain- and loss-of-function experiments suggest that the p38 and JNK pathways are negative regulators of the hypertrophic response.



