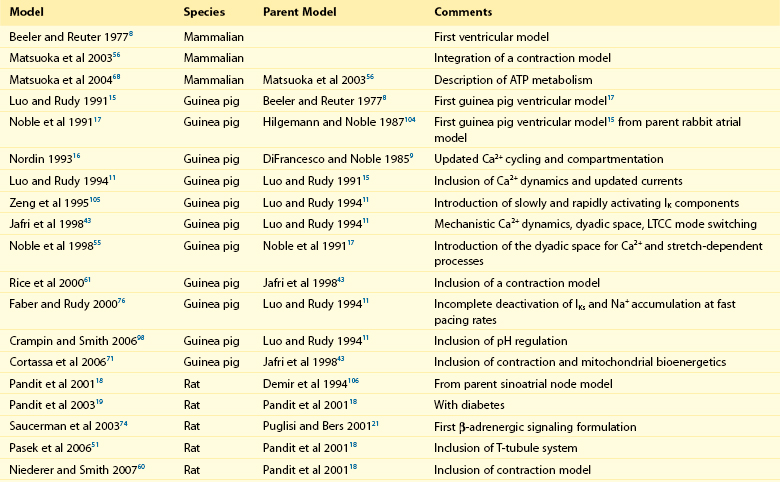
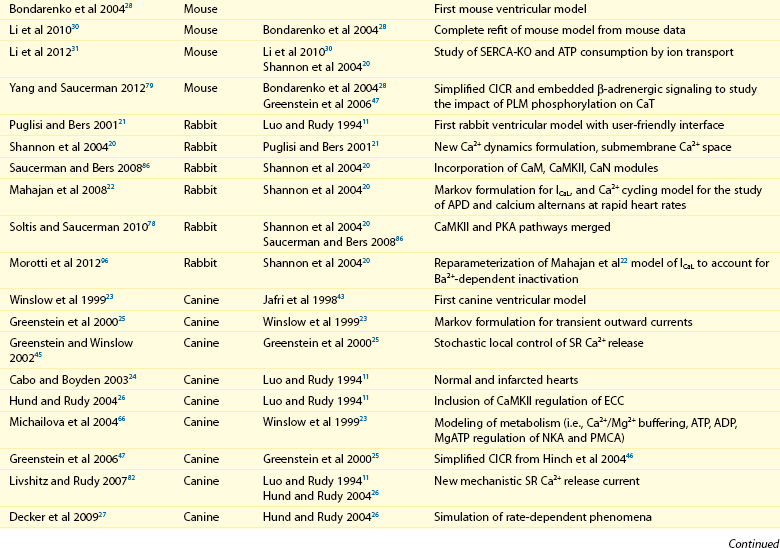
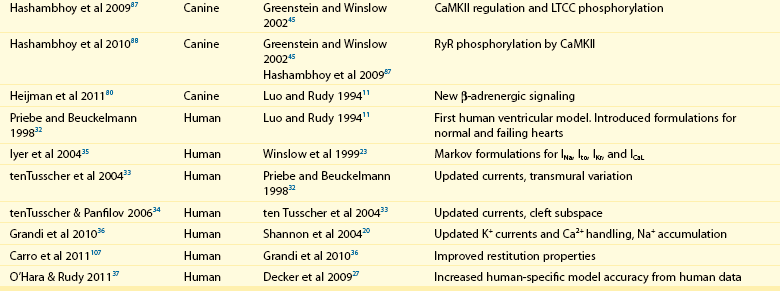
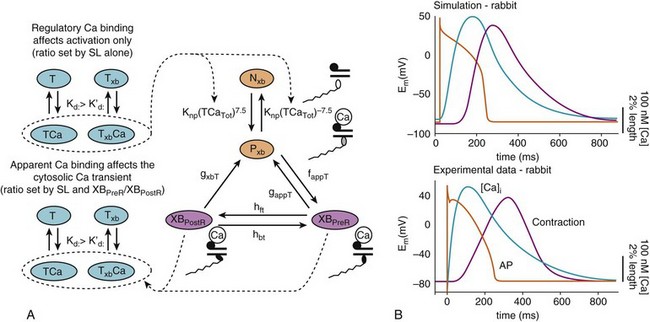
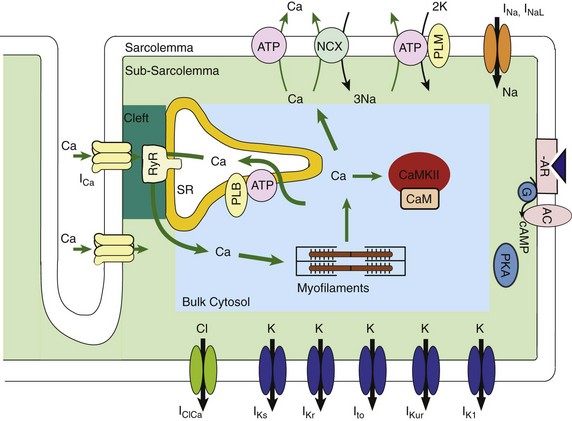
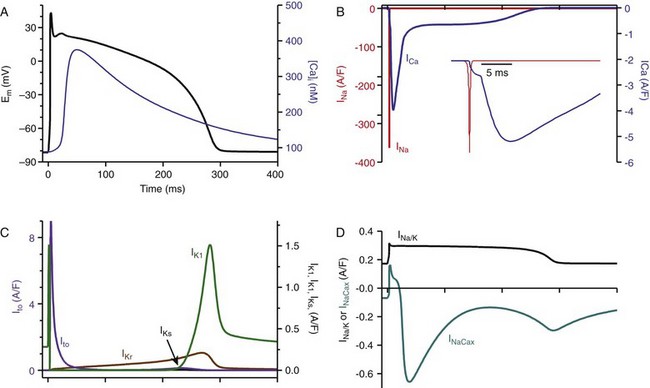
32 The action potential (AP) is a transient depolarization of the cell membrane that emerges from the dynamic behavior of a diverse population of membrane ion channels (depicted in Figure 32-1). A prototypical ventricular myocyte AP is shown in Figure 32-2, A (black). The AP exhibits a steep upstroke, followed by a sustained slowly decaying plateau phase, which eventually gives way to repolarization. Below the AP are shown the associated depolarizing currents, which are carried by inward Na+ and Ca2+ currents. Under physiological conditions, the Na+ current (INa) activates rapidly, producing the AP upstroke, and then inactivates completely (Figure 32-2, B, red). The L-type Ca2+ current (ICaL) inactivates more slowly (Figure 32-2, B, blue), and less completely, allowing for the inward Ca2+ current to maintain the plateau phase of the AP. The influx of Ca2+ via ICaL triggers the release of Ca2+ from the sarcoplasmic reticulum (SR), the subcellular organelle that stores and releases the majority of Ca2+ during each heartbeat.1 This event is known as Ca2+-induced Ca2+ release (CICR) and will be discussed in Section II. The SR then actively re-sequesters Ca2+ via the SR Ca2+ adenosine triphosphatase (ATPase) (SERCA), which is the primary mechanism removing Ca2+ from the cytosol to allow relaxation in between heartbeats.2 Throughout this process, Ca2+ is buffered by Ca2+-binding proteins such as calmodulin (CaM) and troponin. The rise in cytosolic Ca2+ (Figure 32-2, A, blue) ultimately leads to cell contraction (see Section III). The intracellular Ca2+ signal also feeds back on the L-type Ca2+ channel (LTCC), mediating inactivation of the current, and therefore plays a role in influencing the AP shape. Furthermore, intracellular Ca2+ regulates a variety of processes including mitochondrial adenosine triphosphate (ATP) production (see Section IV), intracellular signaling (presented in Section V), and gene expression. Figure 32-1 Schematic representation of the processes described in ECC models. Em homeostasis is regulated by a diverse population of ionic currents: INa: fast Na+ current; INaL: late Na+ current; ICaL: L-type Ca2+ current; IK1: inward rectifier K+ current; IKur: ultra-rapid delayed rectifier K+ current; Ito: transient outward K+ current; IKr: rapidly activating delayed rectifier K+ current; IKs: slowly activating delayed rectifier K+ current; and IClCa: Ca2+-activated Cl− current. The depicted compartmentation for Ca2+ (and Na+) signaling, including a cleft subspace for SR Ca2+ release and a submembrane compartment where Ca2+ raises higher versus bulk [Ca2+], was proposed by Shannon et al20 and was applied by Mahajan et al,22 Grandi et al,36 and Li et al31 in rabbit, human, and mouse models, respectively. Ca2+ signaling interacts with CaMKII and PKA signaling pathways, as illustrated (modeled) in Figure 32-4. Figure 32-2 (A) Human ventricular AP and CaT simulated with the Grandi-Pasqualini-Bers model.36 (B) Na+ and Ca2+ currents (inset shows the different activation and decay times), (C) K+ currents, and (D) Na+/K+ pump and Na+/Ca2+ exchange currents during an AP. Various types of K+ channels (Figure 32-2, C) drive cell membrane repolarization. The transient outward currents (Ito and IClCa), carried by K+ and Cl− respectively, determine the notch that follows the upstroke, and components of the delayed rectifier K+ current (IKr and IKs) contribute to AP repolarization. The inward rectifier current (IK1) maintains and stabilizes the resting potential. Other important players in shaping properties of the cardiac AP are the Na+/K+ pump (NKA), which generates an outward current by extruding 3 Na+ ions and importing 2 K+ ions on each cycle, and the Na+/Ca2+ exchanger (NCX), which mostly operates in the Ca2+-extrusion mode and yields a net inward charge movement when 1 Ca2+ ion is exchanged for 3 Na+ ions (Figure 32-2, D). The plasma membrane Ca2+ ATPase (PMCA) also extrudes Ca2+ from the cell and finely regulates cytosolic Ca2+ levels. Hodgkin and Huxley laid the foundation for the use of integrative models in biology describing both voltage clamp measurements of membrane currents and an integrative AP model of the squid giant axon.3,4 Their modeling approach, which postulated that gating mechanisms regulated membrane permeability, whereby distinct entities (i.e., gates) controlled the flux of both Na+ and K+ ions, has served to this day as a paradigm for a quantitative description of cell membrane excitability. In the early 1960s, Denis Noble presented the first computational models of a cardiac myocyte5,6 and addressed the issue of whether Na+ and K+ current descriptions similar to those of Hodgkin and Huxley could be employed to account for the long AP plateau in Purkinje fibers. Modifications to Na+ and K+ current kinetics were able to generate a plateau, which was supported by the inward Na+ current. It is now known that Ca2+ currents, which had not yet been discovered and were later introduced in the McAllister-Noble-Tsien model,7 mainly support the AP plateau. Beeler and Reuter extended the latter model to develop the first computational ventricular myocyte model.8 Their work introduced a time-varying intracellular Ca2+ concentration and reinforced the idea that a balance between K+ and Ca2+ currents maintains the AP plateau, and repolarization is regulated by activation of K+ and inactivation of Ca2+ currents. Aspects of intracellular Ca2+ handling were introduced in the DiFrancesco-Noble Purkinje cell,9 which constituted the basis of all subsequent models of the cardiac myocyte. One of the landmarks in the development of models of ventricular electrophysiology was the formulation of the dynamic Luo-Rudy ventricular cell model.10,11 Important features of this model were the inclusion of the intracellular SR compartment, time-varying intracellular ion concentrations, and ion pumps and exchangers. The continued interactive iteration between experiments and simulations has led to improved mechanistic insights into cardiac myocyte electrophysiology, as was recently reviewed.12 Table 32-1 summarizes the existing models of the ventricular myocyte, based on previous classifications.12–14 Although most of the early models generically integrated experimental data from mammalian hearts (mostly guinea pig11,15–17), by the mid-1990s, electrophysiological studies had shown species differences in AP waveforms and ionic currents (e.g., mouse and rat APs lack a plateau phase), but exhibited rapid repolarization and very short AP duration (APD) if compared with the prominent plateau phase and long AP seen in humans, rabbit, guinea pig, and dog. Thus, the emphasis shifted to more detailed models on the basis of data obtained from isolated cells from one particular species (see Table 32-1). Rat ventricular cell models have been developed,18,19 as well as rabbit20–22 and dog.23–27 Mouse models28–31 are becoming increasingly important, as genetic manipulation in mice has proven to be a powerful tool for studying the physiological effects of gene mutations, knockouts, and overexpression. The wealth of experimental data that can be measured in these mouse models cannot be obtained in humans, but integrative models provide a systematic framework to make inferences concerning the effects expected in other species and in the clinical setting (see caveat later). The first human ventricular cell model was published by Priebe and Beuckelmann32 and was largely based on the dynamic Luo-Rudy model,11 in which formulations for the major ionic currents were adjusted to the data (limited at that time) available for human ventricular cells. The tenTusscher et al33 model and an updated version34 included many reformulated currents and recapitulated several electrophysiological phenomena. The Iyer et al35 model addressed whole-cell Ca2+ homeostasis carefully. The most relevant ionic currents were based almost entirely on data from human channels expressed in non-myocytes and formulated with Markovian chains, which made this model much more complex than those previously described. The Grandi-Pasqualini-Bers model36 included K+ current reformulations using undiseased human data, provided an accurate description of Ca2+ and Na+ handling in the human ventricular myocyte, and accurately reproduced diverse aspects of excitation-contraction coupling (ECC), including the contribution of various K+ currents to repolarization reserve, the phenomenon of reverse-rate dependence of APD upon K+ current block, and feedback on the effects of changes in Na+ levels on APD and Ca2+ handling. The O’Hara and Rudy model37 included new measurements for ICaL, K+, and NCX currents from undiseased human ventricle, and included the effects of Ca2+/CaM-dependent protein kinase II (CaMKII) on ionic currents and Ca2+ cycling. This model also reproduced early afterdepolarizations (EADs) and alternans. Species-specific models provide a useful platform for investigating species differences (e.g., with respect to arrhythmia and drug response). Comparison of AP repolarization, rate-dependent behavior, and drug response in human, dog, and guinea pig demonstrated major species differences and suggested that great caution should be taken when attempts are made to extrapolate results from nonhuman species to human cellular electrophysiology.38 The opening of LTCCs upon membrane potential (Em) depolarization allows Ca2+ to flow down its electrochemical gradient into the dyadic subspace and trigger the opening of the ryanodine receptors (RyRs) that release Ca2+ from the SR via CICR. At more negative Em, fewer Ca2+ channels open, but the unitary flux is larger (because of the larger driving force), causing more efficient Ca2+-induced Ca2+ release and higher ECC gain (ratio of Ca2+ released from the SR to the Ca2+ trigger via LTCCs). Gain at negative Em is high, in part because a single Ca2+ channel opening may trigger junctional release, but as more channels are recruited (e.g., approaching 0 mV), greater trigger redundancy of LTCC opening (and a consequently large denominator in the gain equation) is seen per release unit.39 At increasingly positive Em (where unitary currents are small), multiple openings are needed to ensure RyR opening, which results in lower gain. The Ca2+ release flux is a smooth, continuous function of trigger influx—a behavior observed originally by Fabiato40 and known as graded Ca2+ release. Whereas the dynamic Luo-Rudy model11 generated APs using detailed kinetic descriptions of membrane currents, the Ca2+ subsystem was represented by a phenomenological model mimicking the process of CICR, but unable to capture the biophysical details involved. Many advances have resulted from improved descriptions of intracellular Ca2+ handling (see recent reviews41,42). In 1998, Jafri et al incorporated in a model of the guinea pig ventricular myocyte43 mechanistic Markov models for both LTCCs and RyRs and a restricted subspace (a single compartment representing the total volume of all dyads) into which all Ca2+ fluxes through these channels are directed and [Ca2+] increases faster and higher than measured in the bulk cytosolic compartment. Winslow et al23 adapted this Ca2+ subsystem in a canine model. These “common pool” models, whereby sarcolemmal Ca2+ influx enters the same Ca2+ pool into which SR Ca2+ is released and by which it is regulated, cannot reproduce both high gain and graded release.44 Most models have circumvented this problem by introducing a dependence of the SR Ca2+ flux on LTCC influx or Em, which removes the positive feedback effect inherent to common pool models, resulting in nonphysiological regenerative, all-or-none rather than graded, Ca2+ release. Graded release arises from local stochastic interactions between LTCCs and RyRs in thousands of Ca2+-release units; this process is called local control of Ca2+ release. Several computational models have been developed to investigate properties of local Ca2+ release, but only in 2002, Greenstein and Winslow45 developed a comprehensive model of the ventricular myocyte based on the theory of local control of SR Ca2+ release, which recapitulated several behaviors from the single channel to the whole-cell level. A simplified version of local control of CICR describing the (deterministic) ensemble behavior of release units was developed by Hinch et al46 and was incorporated in a less computationally expensive ECC model.47 Shannon et al20 introduced a subsarcolemmal Ca2+ compartment (see Figure 32-1) that was based on experiments suggesting that the Na+/Ca2+ exchanger senses elevated Ca2+ levels (compared with the bulk [Ca2+]i), and used a model of RyR that included regulation by both cytosolic and luminal [Ca2+]. The model reproduced the steep relationship between SR Ca2+ load and release, and conferred importance to the (partial) contribution of SR depletion in the regulation of SR Ca2+-release dynamics and termination. This Ca2+ subsystem has been adapted in the Grandi-Pasqualini-Bers36 human ventricular myocyte model. Localization and compartmentation of ion channels and transporters have proven crucial for proper ECC (especially in the dyadic space). It has been suggested that NCX and NKA constitute a molecular complex with ankyrin-B and the inositol 1,4,5-phosphate-operated Ca2+-releasing channel in the T-tubules (distinct from the LCC-RyR complex) that may play an important role in the regulation of local Na+ and Ca2+ concentrations and may possibly modulate CICR. In fact, alterations in ankyrin-B levels can affect local [Na+]i and cellular and SR Ca2+ cycling48,49 and are associated with a broad spectrum of cardiac consequences.50 Other local Ca2+ (and Na+) signals may be relevant to ECC. For example, Pasek et al51 proposed a model of the guinea pig ventricular myocyte that includes a diffusive T-tubule system with a heterogeneous distribution of ion channels between tubular and surface membranes, and predicted that Ca2+ depletion and K+ accumulation in the T-tubule during APs impact ECC. Also, the hypothesis that juxtaposition of mitochondria and dyads may give rise to an additional Ca2+-signaling microdomain regulating mitochondrial ATP production and modulating dyadic Ca2+ concentrations may guide future modeling efforts. Ca2+ released in the dyadic subspace during CICR is then free to diffuse out into the myoplasm and throughout the sarcomere, where it binds to the Ca2+-binding protein troponin in the myofilaments and initiates cell contraction. Not only does Ca2+ activate the myofilaments, but also the Ca2+ transient (CaT) is influenced when developed force is changed. Thus it is important to couple models of cardiac electrophysiology and Ca2+ cycling with models of myofilament activation and crossbridge interactions. However, only a few groups have developed detailed systems models of cooperative myofilament activation, in part because of the complexity of spatially explicit models and the theoretical shortcomings of spatially compressed models.52 The first ventricular myocyte model to couple myocyte ionic currents and Ca2+ handling to myofilament interactions was published by Michailova et al in 199253 and was extended in 1997.54 Noble et al55 studied length- and tension-dependent changes in mechanical and electrophysiological processes by incorporating in the earlier Noble et al model17 a description of force generation. Matsuoka et al56 included the Negroni-Lascano myofilament model57 and recapitulated several aspects of ECC. Niederer and Smith58 integrated the Hunter-McCulloch-ter Keurs myofilament model59 into the Pandit et al rat ventricular cell model,18 and this framework was subsequently used to evaluate proposed mechanisms underlying the slow force response to stretch.60 The Rice-Jafri-Winslow model61 incorporated into the Jafri et al43 guinea pig model a formulation of contraction62 that included a phenomenological representation of cooperative interaction between neighboring troponin/tropomyosin units. The model was used to examine aspects of short-term interval–force relationships in cardiac muscle, and showed that cooperative properties of the myofilaments profoundly affect the developed force. Rice et al have expanded this formulation in a new approximate model of myofilament activation and crossbridge cycling (Figure 32-3, A) that captured many experimentally observed features of cooperative length-dependent thin-filament activation.63 The contraction model was then integrated into the Shannon et al20 rabbit ECC framework, showing its suitability for coupling with existing models of electrophysiology and Ca2+ handling, and the ability to recapitulate common experimental characterization such as cell shortening (Figure 32-3, B). The McCulloch group coupled this model to the canine ECC model64 and found that heterogeneities in ion channel and Ca2+ handling protein expression between epicardial, M-cell, and endocardial myocytes in the dog were sufficient to explain most but not all differences between unloaded shortening twitches measured in canine myocytes, and indicated that variations in crossbridge function may be present.
Models of the Ventricular Action Potential in Health and Disease
Computational Models of the Ventricular Myocyte
The Ventricular Action Potential
Early Canonical Models of the Cardiac AP
Species-Specific and Human Models
Ca2+-Induced Ca2+ Release
Force
Models of the Ventricular Action Potential in Health and Disease