(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
The two opposing highly regulated processes, division (fission) and fusion, determine cell-type-specific mitochondrial morphology, intracellular distribution, and activity. These finely tuned processes modulate a variety of mitochondrial functions allowing living cells to respond properly to frequently changing environmental conditions. Tightly controlled balance between fusion and fission is of particular importance in the high-energy-demanding cells, such as cardiac, skeletal muscle, or neuronal cells. A shift toward fission leads to mitochondrial fragmentation, observed in quiescent cells, while a shift toward fusion results in the formation of the large mitochondrial networks, found in metabolically active cells.
Large dynamin-related proteins, mitofusins (MFN1, MFN2) and optic atrophy 1 (OPA1) protein, constitute the core machinery promoting mitochondrial fusion. MFN1 and MFN2 are localized on the mitochondrial outer membrane (MOM) and mediate MOM fusion, while OPA1, located in the mitochondrial inner membrane (MIM), promotes fusion of the MIM as well as cristae remodeling. A dynamin-related protein 1 (DRP1), fission 1 (FIS1) protein, and mitochondrial fission factor (MFF) are core components of mitochondrial fission machinery in human cells. All three fission proteins are localized in the MOM, while currently very little is known about fission of the MIM.
Impairment of mitochondrial dynamics has been associated with various human disorders. Currently the most characterized mutations in components involved in mitochondrial dynamics have been found in inherited neurodegenerative disorders, such as autosomal dominant optic atrophy, Charcot-Marie-Tooth neuropathy, abnormal brain development, and the Wolf-Hirschhorn syndrome. Defects in mitochondrial dynamics also contribute to the pathogenesis of age-related progressive neurodegenerative disorders, such as Alzheimer’s, Parkinson’s, and Huntington’s diseases.
Although the myocardium is characterized by high levels of the major factors involved in mitochondrial fusion and fission, the role of mitochondrial dynamics in the heart has only begun to be uncovered. Emerging evidence strongly suggests that impairment of balance of mitochondrial fusion and fission can be associated with various cardiovascular diseases, including ischemia-reperfusion injury (IRI), various cardiomyopathies, and heart failure (HF).
Introduction
In the past two decades, the concept of mitochondria as relatively static organelles has significantly changed. It has been demonstrated that the two opposing highly regulated processes, division (fission) and fusion, determine cell-type-specific mitochondrial morphology, intracellular distribution, and activity (Fig. 17.1). Furthermore, mitochondria can move along the cytoskeleton interacting with various intracellular organelles and ensuring region-specific cellular requirements. These finely tuned processes, which have been termed mitochondrial dynamics, modulate a variety of mitochondrial functions allowing living cells to respond properly to frequently changing environmental conditions.
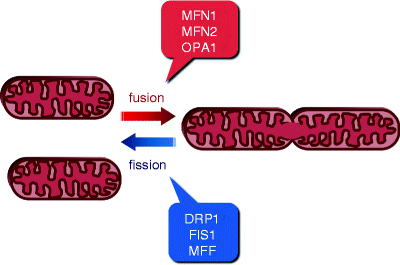
Fig. 17.1
Schematic representation of mitochondrial fusion and fission. The two opposing tightly balanced processes, fusion and fission (division), determine cell-type-specific mitochondrial morphology, intracellular distribution, and activity. Major proteins mediating these processes are shown. Mitofusins 1 and 2 (MFN1 and MFN2, respectively) and optic atrophy protein 1 (OPA1) promote mitochondrial fusion, while dynamin-related protein 1 (DRP1), mitochondrial fission protein 1 (FIS1), and mitochondrial fission factor (MFF) promote mitochondrial fission
Tightly controlled balance between fusion and fission is of particular importance in the high-energy-demanding cells, such as cardiac, skeletal muscle, or neuronal cells. A shift toward fission leads to mitochondrial fragmentation, observed in quiescent cells, while a shift toward fusion results in the formation of the large mitochondrial networks, found in metabolically active cells [1, 2]. Mitochondrial fusion is essential for maintenance and inheritance of mtDNA, transmission of membrane potential, and Ca2+ signaling along mitochondrial networks [1, 3–5]. Mitochondrial fission plays important roles in mitochondrial partitioning during cell division, cytoskeleton-mediated trafficking to energy-demanding intracellular compartments, and removal of damaged mitochondria by mitophagy [1, 5–7]. Moreover, the elongation of mitochondrial tubules has been shown upon differentiation of embryonic stem cells into cardiomyocytes, while mitochondrial fragmentation can contribute to the cytochrome c release and eventually to apoptosis [8, 9].
Evolutionary conserved core components of the mitochondrial fusion and fission machineries have initially been identified in yeast and Drosophila and subsequently in mammalian and human cells [10–13]. In humans, large dynamin-related GTPases, mitofusins 1 and 2 (MFN1 and MFN2, respectively), and optic atrophy protein 1 (OPA1) constitute the core machinery promoting mitochondrial fusion. MFN1 and MFN2 are localized on the mitochondrial outer membrane (MOM) and mediate MOM fusion through interaction with their homologous proteins in adjacent mitochondria. OPA1, located in the mitochondrial inner membrane (MIM), promotes fusion of the MIM as well as cristae remodeling.
A dynamin-related protein 1 (DRP1, also known as DLP1), mitochondrial fission protein 1 (FIS1), and mitochondrial fission factor (MFF) are core components of mitochondrial fission machinery in human cells. All three fission proteins are localized in the MOM, while currently very little is known about fission of the MIM.
Molecular structure of major proteins involved in mitochondrial fusion and fission as well as mechanisms and regulation of these complex processes has been discussed in Chap. 3. In this chapter, the emerging role of mitochondrial dynamics in myocardial and other disorders will be discussed.
Mitochondrial Dynamics in Human Pathology
Animal models have confirmed an essential role of mitochondrial dynamics in cellular function and survival. Mice deficient in MFN1, MFN2, or OPA1 die at early embryonic stages, and downregulation of these factors leads to mitochondrial fragmentation and mitochondrial dysfunction [14–16]. Attenuation of DRP1 using RNAi causes embryonic death in Caernorhabditis elegans [17].
Impairment of mitochondrial fusion and fission has been associated with various human disorders. Energy-demanding neuronal cells are particularly sensitive to alterations in mitochondrial dynamics. Therefore, it is not surprising that currently the most characterized mutations in components involved in mitochondrial dynamics have been found in inherited and age-associated neurodegenerative disorders (Table 17.1).
Table 17.1
Human neurodegenerative disorders caused by impairment of mitochondrial dynamics
Disorder | Affected gene | Affected process | Neuronal defect |
|---|---|---|---|
Autosomal dominant optic atrophy (Kjer’s disease) | Mutations in OPA1 | Impaired mitochondrial fusion | Degeneration of retinal ganglion cells with subsequent atrophy of optic nerve |
Charcot-Marie-Tooth neuropathy type 2A type 4A | Mutations in MFN2 Mutations in GDAP1 | Impaired mitochondrial fusion | Degeneration of peripheral nerves without (type 2A) or with (type 4A) demyelination |
Abnormal brain development | Mutations in DRP1 | Impaired mitochondrial fission | Microcephaly, abnormal brain development, neonatal lethality |
Wolf-Hirschhorn syndrome | Mutations in LETM1 | Impaired mitochondrial dynamics (function unknown) | Growth and mental retardation, congenital hypotonia and heart abnormalities, seizures |
Hereditary early onset Parkinson’s disease | Mutations in PINK1 or PARK2 | Impaired mitochondrial dynamics | Degeneration of dopaminergic neurons in the substantia nigra |
Autosomal Dominant Optic Atrophy
Mutations in the OPA1 gene were identified in 2000 in patients with autosomal dominant optic atrophy (ADOA) [18, 19]. Five loci, OPA1 to OPA5, have been associated with the disease; mutations in OPA1 are the most common cause of ADOA [20]. This heterogeneous disease is the most common form of inherited childhood blindness and characterized by progressive loss of vision due to a primary degeneration of retinal ganglion cells with subsequent atrophy of the optic nerve [21, 22].
Among 117 OPA1 mutations, identified currently, ∼41% have been found in GTPase domain, 33% in the middle domain, 14% in GTPase effector domain (GED), and 12% in the N-terminal region of OPA1 (Fig. 17.2) [23]. The majority of these mutations represent substitutions causing premature truncation of the OPA1 protein. Consistently, skin fibroblasts harboring OPA1 mutations display defects in mitochondrial fusion and OXPHOS [24]. Moreover, inhibition of OPA1 using RNAi has resulted in mitochondrial fragmentations, perturbed cristae structure, decreased ΔΨm and mitochondrial respiration, and elevated cell death [25–28].
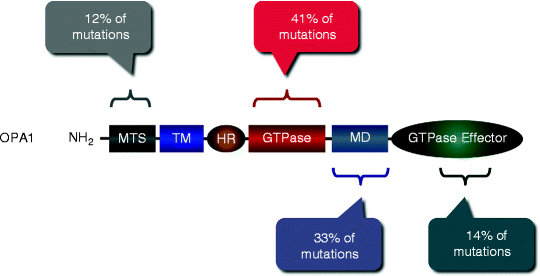
Fig. 17.2
Mutations in the OPA1 gene associated with autosomal dominant optic atrophy. More than 117 OPA1 mutations have been identified; their locations and relative abundance along with OPA1 domains are schematically shown. MTS mitochondrial targeting sequence; TM transmembrane domain; HR heptad repeat region; GTPase GTPase domain; MD middle domain; and GTPase effector domain
Charcot-Marie-Tooth Neuropathy
Charcot-Marie-Tooth disease (CMT) is the most common hereditary neuropathy, affecting approximately 1 in 2,500 individuals [29]. CMT is a heterogeneous hereditary disorder subdivided into demyelinating (CMT1, CMT3, and CMT4) and axonal (CMT2) forms. Demyelinating forms are characterized by axonal de- and remyelination, whereas axonal form, CMT2, is associated with chronic degeneration of peripheral nerves without demyelination, resulting in distal sensory and motor nerve impairments [30, 31].
Charcot-Marie-Tooth Neuropathy Type 2A
Missense mutations in the MFN2 gene have been identified to be responsible for CMT type 2A (CMT2A) [32]. Currently, more than 40 MFN2 mutations have been described in patients with CMT2A [32–37]. Similar to OPA1 mutations, most of MFN2 mutations (>50%) have been detected in the GTPase domain (amino acid residues 99–310; Fig. 17.3). MFN2 mutants display significant functional heterogeneity: some mutations cause gain of function, while others result in loss of function [11, 38, 39].
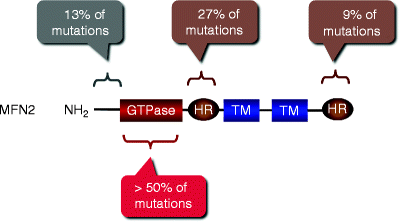
Fig. 17.3
Mutations in the MFN2 gene associated with Charcot-Marie-Tooth neuropathy type 2A. Locations and relative abundance of MFN2 mutations along with MFN2 domains are schematically shown. GTPase GTPase domain; HR heptad repeat region; TM transmembrane domain
Mice expressing Mfn2 mutant with clinically relevant substitution in the GTPase domain (Threonine-105 > Methionine-105) display abnormal mitochondrial dynamics and phenotype similar to clinical symptoms observed in CMT2A [40]. Several other clinically relevant Mfn2 mutants have been demonstrated to cause defects in mitochondrial fusion or mobility [38, 39]. Moreover, cerebellum-specific Mfn2-defficient mice display abnormal mitochondrial arrangement in Purkinje cells and neurodegeneration [41]. Finally, physical interaction between MFN1 and MFN2 may explain compensation of loss of the MFN2 function and therefore the absence of impairments in mitochondrial dynamics in some CMT2A patients [11, 42, 43].
Charcot-Marie-Tooth Neuropathy Type 4A
In 2002, mutations in the gene encoding ganglioside-induced differentiation-associated protein 1 (GDAP1) were identified in patient with demyelinating form of CMT, CMT4A [44, 45]. GDAP1 is a transmembrane protein anchored by its C-tail in the MOM, which appears to be involved in the mitochondrial fission. Consistent with this role, the loss of GDAP1 results in elongated mitochondria, whereas its overexpression facilitates DRP1-medited mitochondrial fragmentation. [46, 47] Several mutations in the GDAP1gene that affect fission activity have been identified in patients with CMT4A axonopathy; however, more studies are needed to elucidate a genotype-phenotype relationship in patients with CMT4A.
Abnormal Brain Development
A heterozygous missense mutation in the DRP1gene, encoding the core fission protein DRP1, has been identified in a newborn girl with microcephaly and abnormal brain development [48]. This mutation was lethal causing the patient death at the age of 37 days. The mutation represents a substitution “Alanine-395 > Aspartic acid-395” (A395D), mapped to a conserved amino acid residue in the middle region of DRP1. Importantly, upon expression in fibroblasts, the A395D DRP1 mutant acts as a dominant form attenuating mitochondrial and peroxisomal fission [48].
The Wolf-Hirschhorn Syndrome
The Wolf-Hirschhorn syndrome (WHS) is a complex hereditary disorder resulted from the deletion of the distal short arm regions of chromosome 4, affecting approximately 1 in 50,000 individuals [49]. Affected patients have distinct facial appearance and display growth and mental retardation, congenital hypotonia and heart abnormalities, midline defects, and seizures. Although a 165-kb WHS critical region has been mapped, the number of genes, which contributes to the disorder, is currently unknown [50]. The gene encoding leucine zipper-EF-hand containing transmembrane protein 1 (LETM1) is mapped approximately 80 kb distal to the WHS critical region [51]. Importantly, LETM1 has been found to be deleted in all WHS patients, displaying all characteristic symptoms of the disorder. LETM1 is a transmembrane protein, which is located within the MIM and appears to be involved in the maintenance of mitochondrial homeostasis and dynamics [52–54]. This finding represents an example of identifying a new factor playing a role in mitochondrial dynamics; however, the mechanisms underlying LETM1 function remain to be elucidated.
Obesity and Type 2 Diabetes
Obesity and type 2 diabetes are characterized by impaired ability of the muscles to switch between glucose and lipid oxidation depending on energy demands. Upon insulin stimulation, subjects with these conditions do not switch from lipid to glucose oxidation, i.e., display decreased metabolic flexibility [55–57]. As mitochondria play a key role in these oxidative pathways, it is not surprising that the impairment of mitochondrial morphology and function has been detected in obese and type 2 diabetic patients.
A significant downregulation of Mfn2 expression has been shown in skeletal muscles in a rat model for obesity [58]. This downregulation has been associated with the decreased extent of the mitochondrial network suggesting alteration of mitochondrial dynamics. Moreover, a decreased expression of some subunits of OXPHOS complexes in this model is very similar to that described in Mfn2 −/− -deficient muscle cells [59]. Consistently, reduced levels of both MFN2 mRNA and MFN2 protein have been found in skeletal muscles of both obese males and females [58, 60]. MFN2 expression has also been reduced in skeletal muscles of both obese and nonobese type 2 diabetic patients compared to healthy subjects [60].
Finally, impairment of mitochondrial dynamics has been demonstrated to contribute to the pathogenesis of age-related progressive neurodegenerative diseases, such as Alzheimer’s, Parkinson’s, and Huntington’s diseases [11, 13, 61–64]. Loss-of-function mutations in the two genes, PINK1 and PARK2, encoding PTEN-induced putative kinase 1(PINK1) and Parkin (PARK2), respectively, are responsible for early onset Parkinson’s disease. PINK1 is localized and stabilized in mitochondria with decreased ΔΨm to recruit E3 ubiquitin-protein ligase Parkin to damaged mitochondria and to direct them for destruction by mitophagy [65, 66]. Moreover, the PINK1-Parkin complex promotes ubiquitination of the core component mitochondrial fusion, MFN2. [67] Intriguingly, in humans, PINK1 and Parkin act as pro-fusion factors and their loss stimulates mitochondrial fragmentation, whereas, in Drosophila, they play a pro-fission role [68, 69]. In addition, PINK1 interacts with the mitochondrial transport complex, suggesting the involvement of mitochondrial trafficking in Parkinson’s disease [70]. However, precise role of mitochondrial dynamics in the pathogenesis of age-associated neurodegenerative diseases remains to be elucidated.
Mitochondrial Dynamics in the Normal and Failing Heart
It has been found that high-energy-demanding tissues, such as myocardial and skeletal muscles and brain, have fused interconnected mitochondria with densely packed cristae, whereas tissues with lower energy demands, such as liver, have mainly fragmented mitochondria with less tightly packed cristae [71, 72].
In mammalian cardiomyocytes, mitochondria are tightly packed between the sarcomere myofibrils or between the myofibrils and the plasma membrane or clustered nearby the nucleus. Accordingly, these subpopulations of mitochondria are referred to as intermyofibrillar mitochondria (IMFM), subsarcolemmal mitochondria (SSM), or perinuclear mitochondria (PNM), respectively (see Chap. 3) [73–75]. This specific arrangement of mitochondria creates significant technical difficulties in imaging of mitochondrial dynamics in living adult cardiomyocytes and explains the lack of direct evidence of cardiac mitochondrial fusion or fission determined by confocal microscopy [12, 76]. Although, in contrast to confocal microscopy, electron microscopy cannot visualize directly mitochondrial dynamics, this highly resolving technique has recently been used to study this complex process in cardiac cells. Recent electron microscopy studies strongly suggest that cardiac SSM and PNM undergo fusion and fission as well as translocation while IMFM appear to go through these events more rarely [76]. Importantly, with the use of transmission and three-dimensional scanning electron microscopy, alterations in mitochondrial dynamics have been detected in patients with dilated and mitochondrial cardiomyopathy and myocardial hibernation [77–79].
Another structural feature of intracellular arrangement of mitochondria in cardiomyocytes is their close association with the sarcoplasmic reticulum (SR) [80]. Such localization allows efficient link between high Ca2+ microdomains and energy generation during excitation-contraction coupling [81–84]. It should be noted that Ca2+ plays an essential role in intricate regulation of mitochondrial dynamics [85–87]. Two potential pathways of Ca2+-mediated regulation of mitochondrial dynamics have been suggested. Ca2+ overload can induce calcineurin-mediated activation of fission protein DRP1, while in an alternative pathway, DRP1 is activated through Ca2+-induced CaMKIα [88, 89]. In both scenarios, activated DRP1 translocates to mitochondria to promote mitochondrial fission. In addition, fusion protein MFN2 tethers mitochondria to the endoplasmic reticulum, and this tethering is essential to the control of mitochondrial Ca2+ uptake [90].
Consistent with the essential role of Ca2+ signaling in cardiac mitochondrial dynamics, it has recently been shown that Ca2+ overload induces mitochondrial fragmentation in neonatal and adult rat ventricular cardiomyocytes [91]. Importantly, Ca2+-induced mitochondrial remodeling can be largely prevented by the transfection with a dominant-negative mutant of DRP1 (DRP1K38A). Taken together, these findings demonstrate complex interplay between mitochondrial function and dynamics and Ca2+ signaling in cardiomyocytes. However, the precise mechanisms of these linkages are largely unknown.
Over 40 years ago, it was found that hypoxia induces mitochondrial rearrangement leading to the formation of gigantic mitochondria in perfused rat hearts [92]. However, role of mitochondrial dynamics in the heart has only recently begun to be addressed, and thus, many critical questions await definitive answers. More recently, mitochondrial dynamics has been demonstrated in neonatal cardiomyocytes and cultured cells of cardiac lineage in various experimental settings [91, 93–95]. These cells express cardiac-specific factors, but they have less tightly packed mitochondria due to less well-organized sarcomeres compared to adult cardiomyocytes.
Importantly, the myocardium is characterized by high levels of the major factors, which are involved in mitochondrial fusion and fission [58, 96–98]. Northern and Western blot analysis of MFN1 and MFN2 expression in human heart, brain, skeletal muscle, liver, lung, kidney, pancreas, and placenta has revealed their highest levels in the heart [99]. Four OPA1 splice variant transcripts, resulting in six posttranslationally modified protein isoforms, have been detected in adult murine hearts [100]. High levels of fission proteins FIS1 and DRP1 have also been found in adult human heart, skeletal muscle, and brain [96, 101].
Differentiation of non-beating embryonic stem cells into contracting cardiomyocytes is an energy-demanding process. Consistently, cardiomyocyte differentiation has been associated with a dramatic change in mitochondrial morphology from fragmented mitochondria to interconnected mitochondrial network [8]. Upregulation of the fusion protein MFN2 accompanying downregulation of the fission factor DRP1 appears to be responsible for this morphological transformation [8].
Emerging evidence strongly suggests that impairment of balance of mitochondrial fusion and fission can be associated with various cardiovascular diseases, including ischemia/reperfusion injury (IRI), various cardiomyopathies, and heart failure (HF) (Table 17.2).
Table 17.2
Cardiac disorders associated with impaired mitochondrial dynamics
Cardiac condition | Cell type | Affected process | Molecular defect |
|---|---|---|---|
Ischemia-reperfusion injury | Mouse atrial-derived cardiac cell line HL-1 Adult heart | Mitochondrial fission | DRP1 targeting to mitochondria. Its inhibition by Mdivi-1 prevents mitochondrial fragmentation and apoptosis |
Cardiomyopathy | Adult heart | Mitochondrial fusion | Downregulation of OPA1 Python mutation in the murine Drp1 gene |
Diabetic heart | Mouse coronary endothelial cells and adult heart | Mitochondrial fission | Elevated DRP1 levels and reduced OPA1 levels No changes in MFN1, MFN2, and FIS1 levels |
Ischemia/Reperfusion Injury
Using a mouse atrial-derived cardiac cell line HL-1 and a cell-based model of myocardial IRI, extensive mitochondrial fragmentation has been shown [102]. The changes in mitochondrial morphology have been followed by translocation of pro-apoptotic Bcl-2 proteins to mitochondria. Intriguingly, mitochondrial fragmentation has been reversed by the addition of the p38MAPK inhibitor SB203580 at reperfusion, suggesting that p38MAPK signaling is involved in the IRI-induced mitochondrial remodeling [102].
Ong et al. [95] have demonstrated in adult mouse heart the fragmentation of IMFM caused by a 20-min episode of mild ischemia. Furthermore, exploiting the HL-1-based model of IRI, they have shown that ischemia-induced mitochondrial fragmentation is associated with the recruitment of fission protein DRP1 to mitochondria. Importantly, overexpression of a dominant-negative mutant of DRP1 (DRP1K38A) or the fusion proteins MFN1 or MFN2 has prevented ischemia-induced mitochondrial fragmentation and protected these cells from cell death [95].
In contrast, overexpression of the fission factor FIS1 has stimulated cell death during ischemia. Consistently, Mdivi-1, a pharmacological DRP1 inhibitor, has prevented mitochondrial depolarization, fragmentation, and ischemia-induced cell death in both HL-1 cells and adult cardiomyocytes [95, 103]. In addition, inhibition of DRP1-mediated fission by the Mdivi-1 has cardioprotective effect reducing significantly infarction size after ischemia/reperfusion [95]. Although the precise mechanisms of ischemia-induced mitochondrial fragmentation remain yet to be determined, authors have hypothesized that increased ROS production and Ca2+ overload may contribute to this process. In agreement with this hypothesis, in another cell type, renotubular cells, it has been shown that ischemia-induced mitochondrial fragmentation could be prevented by pretreatment with an antioxidant [104].
Although above data suggest that prevention of mitochondrial fragmentation can be cardioprotective, excessive elongation of cardiac mitochondria is detrimental to cardiomyocytes. It has been demonstrated that mutations in DRP1 have led to interconnected, elongated mitochondria, reduced level of OXPHOS, and caused cardiomyopathy (see below) [105].
Cardiomyopathies
Significant changes in mitochondrial arrangement within cardiomyocytes, suggesting impairment of mitochondrial dynamics, have been found in various cardiomyopathies. In canine models of chronic HF, the increased number of small mitochondria with decreased matrix density has been observed, while cardiac hypertrophy in rats has led to the enlargement of cardiac mitochondria [106, 107]. In dilated cardiomyopathy and myocardial hibernation, fragmentation of the mitochondrial network, resembling that observed during IRI, has also been found [77, 79].
Similar mitochondrial changes have been detected in skeletal muscles from patients suffering from mitochondrial myopathies and in mouse model of mitochondrial cardiomyopathy [108]. Importantly, mitochondrial fragmentation has been associated with dissipation of the mitochondrial membrane potential and a decrease in OPA1 due to its proteolytic processing. Consistently, overexpression of OPA1 has prevented fragmentation of mitochondria [108]. This finding suggests that mitochondrial dynamics and fusion protein OPA1 play a critical role in preventing fusion of dysfunctional mitochondria with the functional mitochondrial network.
It has recently been reported that cardiac-specific ablation of Mfn2 has no significant effect on cardiac function resulting in very modest cardiac hypertrophy [109]. However, conditional cardiac-specific deletion of both Mfn1 and Mfn2 has led to cardiac mitochondrial fragmentation and respiratory dysfunction resulting eventually in lethal dilated cardiomyopathy [110]. These recent findings have further confirmed that cardiac function depends on mitochondrial fusion as it has previously been demonstrated in murine brain and skeletal muscle [41, 111].
Idiopathic dilated cardiomyopathy (DCM) causes HF with a prevalence of 36 per 100,000 individuals in the United States [112]. Although a number of DCM-related mutations, which affect sarcomeric and cytoskeletal integrity, contractile dynamics, and Ca2+ homeostasis, have been found during the past decade, they account for a minority of the cases suggesting that many additional genes, implicated in the pathogenesis of the disease, remain to be determined [113, 114]. Importantly, in patients suffering from DCM, increased number of gigantic mitochondria, containing lower density matrix, has been reported [115]. Earlier, similar formation of giant mitochondria has been observed in rat myocardium under experimental hypoxia [92].
Recently, the first direct demonstration that a mutation in the mouse mitochondrial fission gene Drp1 (also known as Dnm1l) results in cardiomyopathy has been reported [105]. The “Cysteine-452 > Phenylalanine-452” (C452F) mutation, called Python, in a highly conserved region of the middle domain of Drp1 alters its self-assembly into higher-order oligomers, which are essential for Drp1 fission function [105, 116, 117]. Homozygous Python mutation is embryonically lethal, whereas heterozygous Python hearts display reduced mitochondrial ATP production. Furthermore, heterozygous Python fibroblasts contain abnormal mitochondria and peroxisomes [105]. Although it is currently unclear why the C452F Drp1 mutant impacts specifically heart function, this data strongly suggest that imbalance of mitochondrial fusion and fission is implicated in the pathogenesis of cardiomyopathy.
Heart Failure
HF is characterized by significant reduction of mitochondrial respiration accompanied with increased ROS generation. Mitochondrial dysfunction during HF has been shown to be associated with disorganization and fragmentation of cardiac mitochondria similar to those observed in various cardiomyopathies [118]. Authors have also demonstrated a significant decrease in levels of the fusion factor OPA1 in failing hearts, while levels of MFN1, MFN2, DRP1, or FIS1 have not been changed. Moreover, simulated ischemia in cardiac myogenic H9C2 cells has induced a similar decrease in OPA1 levels associated with appearance of disorganized small fragmented mitochondria. Interestingly, opening of mitochondrial permeability transition pore (MPTP) appears to affect OPA1 levels as the pretreatment of cells with MPTP inhibitor cyclosporin A (CsA) has attenuated ischemia-induced downregulation of OPA1 [118]. Although OPA1 overexpression has induced mitochondrial fusion, it has not protected H9C2 cells against ischemia-induced cell death [118]. It has been suggested that OPA1 overexpression at low to moderate levels can cause fusion and be cell protective, while it can lead to fission and cell death at higher levels [119]. As it has been noted above, OPA1 can also play a cardioprotective role contributing to the degradation of dysfunctional mitochondria in failing hearts [108].
Apoptosis of cardiomyocytes is a critical determinant in the development of HF. Mitochondrial fragmentation induced by apoptotic stimuli suggests a link between mitochondrial dynamics and apoptosis [120–122]. Involvement of mitochondrial fission machinery in ceramide-induced apoptosis has been demonstrated in rat cardiomyocytes [97]. Mitochondrial fragmentation, associated with the loss of membrane integrity, results in release of pro-apoptotic factors, such as cytochrome c, APF1, endonuclease G, and Smac/DIABLO, which initiate intrinsic apoptotic pathway. Activation of pro-apoptotic Bax leads to Bax/Bak-mediated sumoylation of fission protein DRP1, which translocates from the cytosol to mitochondria to promote mitochondrial fission [123].
While involvement of mitochondrial fission machinery in apoptosis could be anticipated, contribution of mitochondrial fusion to the process is less obvious. However, more recently, participation of fusion protein MFN2 in this complex process has also been shown. Using cell-free system of mitochondrial fusion, it has recently been demonstrated that Bax stimulates the process through MFN2, but not MFN1 [124]. MFN2 is a critical regulator of apoptotic response in cardiomyocytes: its levels are significantly increased in response to OS, IRI, or cardiac infarction. Moreover, overexpression of MFN2 attenuates Akt signaling and triggers the mitochondria-mediated apoptotic pathway in both vascular smooth muscle cells and cardiomyocytes [125, 126].
Consistent with pro-apoptotic role of MFN2, its depletion has resulted in increased resistance of cardiac mitochondria to Ca2+-induced MPTP opening, and Mfn2 −/− mouse cardiomyocytes are resistant to apoptosis-inducing stimuli [109]. Accordingly, Mfn2-defficient mice have shown better recovery after post-ischemic reperfusion injury.
It has recently been demonstrated that MFN2 is located not only on mitochondria but also on the membranes of endoplasmic reticulum (ER). Thus, ER-located MFN2 may contribute to regulation of Ca2+ flux from the ER to the adjacent mitochondria [90]. This may represent an additional mechanism underlying pro-apoptotic role of MFN2 in cardiomyocytes [109].
However, the contribution of MFN2 to apoptosis is yet unclear and remains controversial. Not only pro-apoptotic effect of MFN2 but also its anti-apoptotic role has been demonstrated in mammalian cells. MFN2 downregulation has acerbated cytochrome c release in cardiomyocytes during ceramide-induced apoptosis [97]. Consistently, dominant-active MFN2 form has interfered with radical-mediated pro-apoptotic Bax activation, while dominant-negative MFN2 mutant has promoted decrease in ΔΨm and induced mitochondrial fragmentation [99, 127].
Intriguingly, the role of MFN2 in apoptosis of cardiovascular cells appears to be independent of its function in mitochondrial fusion [125, 126].
In cardiomyocytes, attenuation of autophagy, which appears to be cardioprotective, can lead to apoptosis during IRI contributing thereby to the development of HF. Selective autophagic removal of damaged mitochondria, mitophagy, in response to ischemic stress mediates elimination of dysfunctional mitochondria [128, 129]. Although an essential role for mitochondrial dynamics in autophagy has been suggested, the mechanistic link between these processes in the pathogenesis of cardiac disease remains to be determined [7, 130].
Diabetic Heart
Mitochondrial oxidative stress (OS) induced by hyperglycemia is believed to be a main mechanism responsible for increased incidence of CVD in patients with diabetes mellitus [131, 132]. It has been reported that hyperglycemia-induced ROS production causes mitochondrial fragmentation leading eventually to MPTP opening and cell death [133]. Interestingly, in diabetic cardiomyopathy, changes in mitochondrial morphology and function, such as decrease in activity of complexes I and III and elevated ROS generation, have been observed mainly in IMFM [134].
It has recently been shown that coronary endothelial cells isolated from mouse diabetic hearts have more mitochondrial fragmentation compared to hearts from nondiabetic mice. Importantly, this change in mitochondrial morphology has been associated with elevated levels of DRP1 and reduced levels of OPA1, while levels of MFN1, MFN2, and FIS1 have not been changed [135]. Surprisingly, while treatment with an antioxidant has attenuated mitochondrial fragmentation, it has not affected changes in the levels of DRP1 or OPA1. Thus, further investigations are needed to access the role of mitochondrial dynamics in the pathogenesis of diabetic cardiomyopathy.
Taken together, these findings reveal that mitochondrial fusion and fission are complex highly regulated processes implicated in various aspects of cardiac physiology. A tight balance of fusion and fission is critical for the maintenance of mitochondrial homeostasis in the heart. An imbalance in these processes can perturb mitochondrial morphology and function and be detrimental to the myocardium contributing to the pathogenesis of various CVDs, including IRI, cardiomyopathies, HF, and diabetic cardiomyopathy.
Conclusions
Over the past decade, cardiac mitochondria have emerged as critical integrators of energy production, generation of ROS, multiple cell death, and signaling pathways in the constantly contracting heart. Finely tuned processes of mitochondrial dynamics contribute to the multifaceted role, which mitochondria play in myocardial physiology. Impressive progress has recently been achieved in deciphering the complex multiprotein machineries that promote mitochondrial fusion, fission, and trafficking. Growing evidence suggests that defects in the core components of these machineries can result in imbalance of mitochondrial fusion and fission leading eventually to various human disorders.
Presently, the most established causative link between mutations in the genes encoding proteins, which mediate mitochondrial fusion and fission, and pathological conditions has been demonstrated in inherited neurodegenerative disorders. They include autosomal dominant optic atrophy, Charcot-Marie-Tooth neuropathy, abnormal brain development, and the Wolf-Hirschhorn syndrome. In addition, impairment of mitochondrial dynamics has been associated with age-related progressive neurodegenerative disorders, such as Alzheimer’s, Parkinson’s, and Huntington’s diseases. Intriguingly, these pathologies are usually not associated with cardiac dysfunction.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


