(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
Aging is associated with progressive impairment of a variety of vital functions, resulting in an increased vulnerability to environmental challenges and a growing risk of disease and death. Cardiac aging, in particular, reduces cardiac functional reserve and predisposes the heart to stress, serving as one of the major risk factors for cardiovascular diseases (e.g., left ventricular hypertrophy, fibrosis, diastolic dysfunction). Thus, aging contributes to increased cardiovascular mortality in the elderly. During the last two decades, several different molecular pathways involved in the aging process have been revealed, and mitochondria were pointed out as one of the key regulators of longevity. Aging of cardiac tissue is accompanied by accumulation of mitochondrial protein oxidation, increased mitochondrial DNA mutations, deterioration of the respiratory chain function, and changes in mitochondrial biogenesis. Respiratory chain-deficient cells are more susceptible to undergo apoptosis, and increased cell loss is likely to be associated with the age-associated mitochondrial dysfunction.
Introduction
The elderly (>65 years old) account for the majority of patients with ischemic heart disease, congestive heart failure, and atrial fibrillation (70–80%) [1]. Cardiac aging is the slowly progressive age-dependent degeneration and decline in function that make the heart more vulnerable to stress and contribute to increased cardiovascular mortality and morbidity in the elderly. Remodeling of the aging heart involves a significant loss of cardiac myocytes, reactive hypertrophy of the remaining cells, and increased connective tissue. The loss of cardiac myocytes with advanced age is mediated by apoptosis, and mitochondria play a key role in regulating this process.
Several theories have been proposed to explain the fundamental mechanisms mediating age-related diseases and conditions, with the free-radical theory of aging being by far the most popular. According to this theory, reactive oxygen species (ROS) cumulatively damage biological molecules leading to irreversible cell damage and an overall functional decline [2]. The current free-radical theory has been extended to include mitochondria, which increasingly produce ROS with aging. ROS overproduction can subsequently lead to a vicious cycle of increasing levels of mitochondrial DNA (mtDNA) damage and oxidative stress in the cell [3–5].
There is controversy regarding the involvement of increased ROS production and enhanced mitochondrial oxidative stress in the contribution of mtDNA mutations to aging [6, 7]. It is possible that the accumulation of mtDNA mutations with aging may affect cell-signaling pathways that induce cell dysfunction and initiate apoptosis, irrespective of increased ROS production and oxidative stress in mitochondria. Whether mtDNA mutations play a causal role in the aging process is still open to discussion.
Oxidative Stress and Aging
More than five decades ago, Harman [2] proposed the free-radical theory of aging, postulating ROS as a determinant of life span. ROS have damaging effects on various cell components and might drive an age-dependent functional decline of cells, leading to associated degenerative diseases. Although several sources within cells contribute to the oxidative complications, mitochondria produce the majority of ROS during oxidative phosphorylation as a by-product of electron transfer.
Mitochondrial proteins, lipids, and nucleic acids can be at high risk from damage caused by ROS. This has led to the mitochondrial variant of the free-radical theory of aging (reviewed in [8]). According to this theory, mitochondrial ROS attack mitochondrial elements, causing damage of proteins, lipids, and mtDNA, followed by mitochondrial dysfunction and a vicious cycle between increased mitochondrial damage and further increase of ROS production, which can lead to the functional declines of cells and organs and eventually to death.
In several studies, an age-dependent impairment of mitochondrial function has been documented. In particular, the decline in mitochondrial respiratory capacity takes place with age, mostly due to diminished activity of respiratory complexes I and IV. The slower rate of electron transfer in aging mitochondria accelerates mitochondrial superoxide production, leading to a positive feedback between complex I inhibition, mitochondrial ROS production (“ROS-induced ROS release”), protein damages, and mtDNA mutation (reviewed in [9]).
Although passive leakage of electron from the mitochondrial electron transport chain (ETC) has been considered as a major source of oxidative stress in the aging heart, mitochondrial member of the NAD(P)H oxidase family, Nox4, was also considered recently to play an important role in mediating aging [10]. Nox4 has a mitochondrial localization signal-like motif in the N-terminal region and appears to localize in mitochondria in cardiomyocytes [11]. This enzyme produces superoxide anion (O2•−) and hydrogen peroxide, thereby contributing to oxidative stress in mitochondria, induces oxidative damage of mitochondrial proteins [aconitase, components of complex I, mitochondrial permeability transition pore (MPTP)], and causes leakage of O2•− from mitochondria [11, 12]. Mitochondrial expression of this actively producing ROS enzyme in the heart is upregulated by aging.
Heart is an organ with high metabolic demand and rich in mitochondria, and it is very vulnerable to mitochondrial oxidative damage. Aging heart is characterized by mitochondrial bioenergetic dysfunction. This dysfunction increases generation of ROS with subsequent oxidative damage of mitochondrial proteins, which is indicated by significant increase of protein carbonyls in mitochondrial extracts from aged hearts [13].
Harmful effect of ROS in the aging heart includes reaction of superoxide with nitric oxide (NO) that forms highly reactive peroxynitrite impairing mitochondrial respiration irreversibly via tyrosine nitration and inhibition of several subunits of complex I [14, 15], modification of cytochrome c structure and function [16], inhibition of cytochrome c oxidase (complex IV) activity, and inhibition of aconitase (Fig. 13.1) [17]. Oxidative damage of mitochondria in old mice heart results in their abnormal ultrastructure, characterized by disrupted cristae and vacuolation [13].
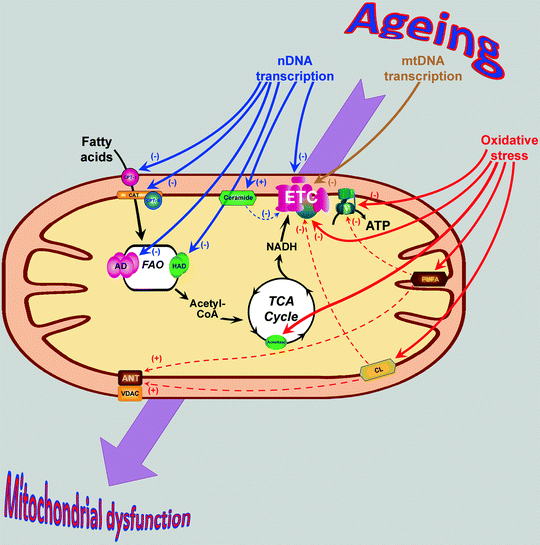
Fig. 13.1
Age-associated changes in mitochondrial proteins and lipids contribute to mitochondrial dysfunction. Aging is associated with decrease of several transcriptional regulators resulting in the decrease of nuclear DNA (nDNA)- and mitochondrial DNA (mtDNA)-encoded mitochondrial proteins (shown by blue and brown arrows, respectively). Additionally, protein components of tricarboxylic acid (TCA) cycle, electron transport chain (ETC), and ATP synthase (V) as well as mitochondrial lipids in aging heart become oxidatively damaged (red arrows). Dashed arrows show secondary effects of the oxidized lipids (see text for details). Changes in the expression/structure of mitochondrial proteins and lipids contribute to the development of mitochondrial dysfunction in senescent myocardium. Abbreviations: AD acyl-CoA dehydrogenase, ANT adenine nucleotide translocator, ATP adenosine-5′-triphosphate, CAT carnitine-acylcarnitine translocase, CL cardiolipin, CoA coenzyme A, CPT carnitine palmitoyltransferase, FAO fatty acid β-oxidation, HAD 3-hydroxyacyl-CoA dehydrogenase, NADH nicotinamide adenine dinucleotide (reduced), PUFA polyunsaturated fatty acid, VDAC voltage-dependent anion channel
Critical role of mitochondrial ROS in cardiac aging has been revealed in experiments of Dai and Rabinovitch [13] on mice overexpressing antioxidant enzyme catalase targeted to the mitochondria (mCAT). They demonstrated that overexpression of mCAT (but not peroxisomal CAT isoform) prolongates life span of mice by 18% [18] and significantly protects from the age-associated cardiac abnormalities (e.g., left ventricular hypertrophy, systolic/diastolic dysfunction, enlargement of left atrium) [13, 19]. Consistently, overexpression of mCAT attenuates age-dependent mitochondrial oxidative damage: It decreases mitochondrial protein carbonyls and protects ultrastructure of mitochondrial cristae [13]. Interestingly, naturally occurring increases in myocardial levels of antioxidative enzymes, such as glutathione peroxidase, glutathione reductase, and MnSOD were also reported in the aging heart [20, 21], which may reflect an adaptive mechanism to neutralize an increased ROS generation. Another mechanism of protection against ROS accumulation in the aging heart involves upregulation of mitochondrial uncoupling proteins (UCPs). UCPs prevent the mitochondrial transmembrane electrochemical potential from being above the value critical for ROS formation by complexes I and III. Importantly, transcription of UCP3 is upregulated in the heart of mice with increased longevity suggesting a protective role of UCPs in the aging heart [22, 23].
Oxidative Damage of Mitochondrial Proteins in Aging Heart
The posttranslational oxidative modifications of proteins appear to be a key mechanism of age-associated oxidative injury. Many studies have shown that accumulation of oxidatively modified molecules is coupled to impairment in mitochondrial function. Declines in activities of the ETC complexes [24, 25] and enzymes of citric acid cycle [26, 27] as well as cytochrome c release from mitochondria [28] of aged heart have been demonstrated and proposed to contribute to dysfunction and death of cardiac cells (Fig. 13.1).
Current data on age-related changes of individual ETC complexes I–IV are contradictory even among studies dealing with the same species. For instance, some studies have reported a significant reduction in complex I activity in hearts of senescent rats [24, 29], whereas other studies have reported no age-related decrease in its activity [30, 31]. Two recent studies have shown small but significant decrease in cardiac complex I activity of old and senescent animals [32, 33]. Similarly, the literature shows decreased activity of complex II in aged rodent myocardium [24, 31], unchanged [29, 33] and even increased [30, 32, 34]. Activities of cardiac complexes III and IV have also been shown to decline [24, 33, 34], remain unchanged, or increase [30–32] during aging of rats.
Such inconsistencies among the studies can be mainly due to differences in the experimental approaches such as mitochondrial subpopulations, electron donors/acceptors used for measurements of complex activities, or differences in aging groups. Nevertheless, regardless of the contradictory data on activity changes of individual ETC complexes, majority of studies demonstrates some age-related impairment in ETC. Such nonuniform inhibition of ETC complexes can result in altered electron flow through the chain, impaired ATP production, and finally in cardiomyocyte dysfunction during aging.
The majority of the literature recognizes that dysfunction of ETC complexes in aging is associated with protein oxidative damage. It has been shown that contents of protein dityrosine, nitrotyrosine, carbonyl, and thiol group are age-related indicating age-related harmful modifications of mitochondrial proteins [31–33, 35, 36]. Besides direct oxidative damage via interaction with ROS, ETC components in aging myocardium may be affected indirectly through modification and inactivation by lipid peroxidation products, such as malondialdehyde (MDA) and 4-hydroxynonenal (HNE) [37–40]. Oxidative protein modifications may be responsible for age-associated destabilization of a native architecture of ETC supercomplexes recently discovered by Gomez et al. [41] (Fig. 13.1).
Mitochondrial ATP synthase (complex V, CV) is not part of the ETC processes but obviously also plays an important role in ATP homeostasis. Some evidences indicate that CV is one of the prime targets for oxidative modification. α chain of CV, for example, becomes HNE-modified with subsequent dissociation from CV [32]. The nitration of another subunit, β-chain, has been shown to be increased dramatically (ten times) in mouse myocardium and correlates directly with the decline in CV function with aging (Fig. 13.1) [32]. Both α and β chains are involved in proton translocation coupled with ATP biosynthesis, and modification of these subunits with aging suggests that they are specific targets of ROS-mediated damage.
Role of Lipids in Age-Related Changes of Cardiac Mitochondria
One of the major features of advanced age is the remodeling of myocardial cell membranes. Age-associated mitochondrial membrane changes include increases in cholesterol, phosphatidylcholine, ω-6 polyunsaturated fatty acids (ω-6 PUFA), and HNE and decreases in ω-3 PUFA and cardiolipin [42]. Moreover, an increase in mitochondrial membrane content of arachidonic acid (20:4 ω-6 PUFA) and a reduction in docosahexaenoic acid (22:6 ω-3 PUFA) have been described in the rat heart with aging [42]. An optimal ratio between ω-6 and ω-3 PUFA is essential for proper functioning of the myocardium, and age-linked changes to cardiac mitochondrial membrane PUFA can influence crucial mitochondrial processes and affect myocardial contractile work, Ca2+ homeostasis, and O2 utilization efficiency and finally reduce cellular capacity to recover from reperfusion injury [43–45].
One important consequence of age-related changes in mitochondrial membranes includes increases in membrane rigidity (i.e., reduced fluidity of the mitochondrial inner membrane [MIM]). Decrease of mitochondrial pool of phosphatidylcholine in comparison with phosphatidylethanolamine in rat heart with the age probably underlies age-related increase of temperature of mitochondrial membrane-lipid phase transition (T t; measure of degree of membrane liquidity) [46–48]. An increased local lipid viscosity of cardiac mitochondrial membrane may alter lipid–protein relationships and thus the activity of intrinsic membrane proteins involved in ion homeostasis, signal transduction, redox reactions, and oxidative phosphorylation. For example, specific lipid microenvironment and fluidity are very important to the function of mitochondrial ATP synthase (complex V): An activity of this enzyme decreases below T t, when membrane lipids are in a liquid-crystalline state [49, 50].
There are also important links between aging and fatty acid unsaturation. Accumulation of PUFA in senescent mitochondria leads to higher levels of lipid peroxidation by increased ROS levels [21, 28], because superoxide reacts with PUFA to generate the highly reactive hydroxyalkenal, HNE [51]. HNE reacts with protein sulfhydryl and thiol groups consequently altering protein conformation affecting thereby numerous enzyme and ion exchange systems [51–54]. In addition to lipid-derived oxidative damage of proteins, damage of DNA appears to be even more important than the pure protein-derived oxidative damage during aging [55]. For instance, protein adducts formed in HNE reactions may adversely modify adenine nucleotide translocator (ANT) and cyclophilin-D to interfere with ΔΨm and ultimately contribute to opening of MPTP (Fig. 13.1) [56]. Detrimental role of PUFA in aging myocardium has been supported by the observations that ω-6 PUFA-rich diets lead to higher levels of lipid peroxidation and DNA double-strand breaks in rat tissues during aging, compared to less oxidizable, MUFA-based diets [57–60]. Interestingly, it has been described that long-lived animals have less unsaturated fatty acids in their mitochondria, which protects them against lipid peroxidation and the negative impact of lipid peroxidation products on proteins and DNA [61, 62].
In recent years, it has been demonstrated that dietary strategy, through changes in membrane-lipid profiles, may help attenuate some age-related deleterious aspects of aging myocardium, such as those related to exacerbated oxidative stress or mitochondrial dysfunction. Diet rich in ω-3 PUFA reverses the age-associated membrane ω-3:ω-6 PUFA imbalance and dysfunctional Ca2+ metabolism, facilitating increased efficiency of mitochondrial energy production and improved tolerance of ischemia and reperfusion [42].
One of the phospholipids, highly enriched in oxidatively sensitive acyl-groups, is cardiolipin. Cardiolipin is an important factor in the regulation of many mitochondrial bioenergetic processes including electron transport, the MIM supermolecular assembly, anion transport, efficient ATP synthesis, binding of cytochrome c, and functioning of the multiple other MIM enzymes [63–65]. This phospholipid is also emerging as an important player in control of the mitochondrial-dependent apoptosis (see section below). Cardiolipin is a unique to mitochondria diphosphatidylglycerol containing 90–95% linoleic acid (18:2) (see Chap. 3 for details) [66, 67]. This phospholipid appears to be an early target for ROS attack due to its high content of unsaturated linoleic acid and its location in the MIM near the site of mitochondrial ROS production. Linoleic acid readily participates in free-radical reactions in the membrane, with the formation of lipid peroxides. The two double bonds can rearrange to a resonance-stabilized conjugated diene, followed by reaction with molecular oxygen to form a lipid hydroperoxide [68]. Alternatively, enzymatic oxidation leads to epoxide formation [69]. Keto-peroxy moieties have been observed under oxidizing conditions in mitochondria [70]. The level and composition of cardiolipin are unaltered, and its oxidation products are not detected in the aged rat heart under normal conditions [71]. Intriguingly, cardiolipin undergoes modification during ischemia in the aged heart [72, 73]. Oxidative damage adds three oxygen atoms to cardiolipin and results in the generation of oxidation product of +48 Da molecular mass [72].
Oxidation of cardiolipin decreases the function of ETC complexes [74, 75], disrupts the bilayer arrangement of the MIM [76, 77], and favors the release of cytochrome c from mitochondria to activate apoptosis [76–78]. In the latter case, cardiolipin under normal conditions localizes cytochrome c at the MIM via both nonionic [79–81] and electrostatic [76] interactions. The oxidative modification of cardiolipin diminishes the affinity of cytochrome c for the MIM [79–81], delocalizing cytochrome c into the intermembrane space [76, 77], the first step leading to cytochrome c release from mitochondria [76]. Cytochrome c, released from mitochondria, in turn activates caspases to induce programmable cell death (Figs. 13.1 and 13.2). Thus, enhanced oxidative damage of cardiolipin within cardiac mitochondria during ischemia may contribute to the mitochondrial-dependent injury that occurs in the aged heart during ischemia and reperfusion [52, 82–87].
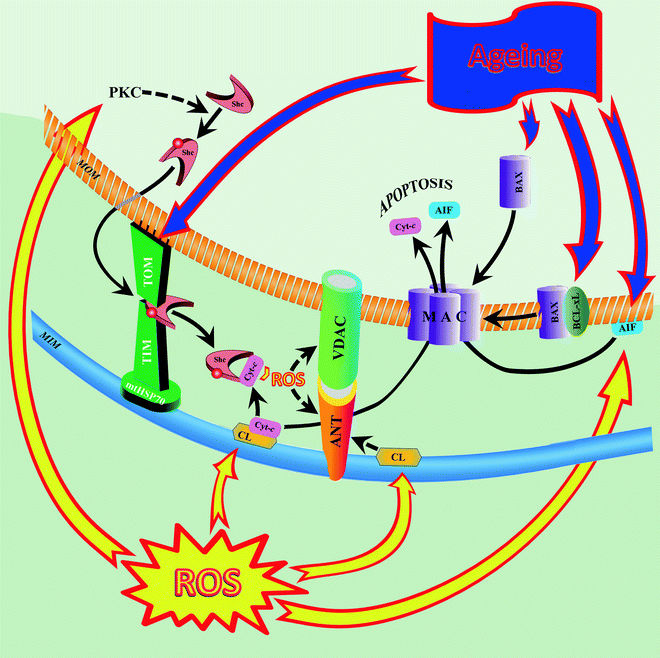
Fig. 13.2
Alterations in aging cardiac mitochondria induce mitochondrially mediated apoptosis. In aging myocardium, increased reactive oxygen species (ROS) activate protein kinase C (PKC) to induce serine-36 phosphorylation of the 66-kDa “human Src homology and collagen” (Shc) protein, allowing transfer of the protein from the cytosol to the mitochondrion. In the mitochondrion, Shc binds to a protein-importing TIM–TOM complex (including mitochondrial 70-kDa heat-shock protein, mtHSP70). Age-related proapoptotic stimuli destabilize the Shc–mtHSP70 complex and lead to the release of Shc. Once activated, Shc oxidizes cytochrome c (Cyt-c) and catalyzes the synthesis of ROS, hydrogen peroxide (H2O2). ROS induces opening of the mitochondrial permeability transition pore, MPTP (two major components of the pore are shown: adenine nucleotide translocator, ANT, and voltage-dependent anion channel, VDAC), with subsequent increase of mitochondrial membrane permeability to ions, solutes, and water, swelling, and disruption of the organelle. Other ROS-dependent events include peroxidation of cardiolipin (CL) and release of apoptosis-inducing factor (AIF) into the cytosol. Peroxidized CL contributes to MPTP opening and detachment of Cyt-c from the inner membrane (first step in the proapoptotic release of Cyt-c from mitochondria). Increased proapoptotic protein Bax and decreased antiapoptotic protein Bcl-xL in the aged myocardium also promote translocation of apoptotic factors Cyt-c and AIF to the cytosol, indicating increased proapoptotic signaling in senescent mitochondria. Abbreviations: MAC mitochondrial apoptosis-induced channel, MIM mitochondrial inner membrane, MOM mitochondrial outer membrane, TIM translocase of the inner mitochondrial membrane, TOM translocase of the outer mitochondrial membrane
Several aspects of age-related changes in mitochondria, such as increased ROS production, oxidative damage, impaired electron transport, and increased apoptosis, are also evident in inflammatory pathologies caused by pro-inflammatory/proapoptotic sphingolipid, ceramide. Cardiac mitochondria normally contain a variety of sphingolipids, including sphingomyelin and ceramide [88], as well as a neutral sphingomyelinase (nSMase), which hydrolyzes sphingomyelin to ceramide [89]. Normal levels of ceramides are vital to maintain optimal membrane fluidity and to modulate many kinases and phosphatases [90]. However, when its levels are elevated, ceramide induces apoptosis, growth arrest [90–92], ROS formation, oxidative stress, and altered energy metabolism [93–95]. Recently Monette et al. [89] discovered that ceramide levels increase by 32% with age in the MIM of rat hearts. An age-related ceramidosis may develop as a result of enhanced hydrolysis of sphingomyelin by nSMase, activity of which doubles with age. Mitochondrial ceramidosis can affect mitochondrial function: For instance, elevated ceramides promote decline in ETC function (Fig. 13.1).
Although the mechanism of nSMase activation in senescent mitochondria is unknown, the role of glutathione (GS) in this process has been suggested. nSMase activity is known to inversely correlate with reduced glutathione (GSH), and V max of enzyme increases in the presence of low GSH levels [96, 97]. The low redox status of GSH is a prerequisite of aged mitochondria [98]. Feeding old rats with the potent antioxidant agent, (R)-α-lipoic acid (LA), restores level of GSH and reduces nSMase activity [89]. Importantly, these effects of LA closely correlate with the decrease in ceramide levels in mitochondria of LA-treated old rats and with restoration of complex IV activity, which otherwise declines with age [89]. Thus, an increased mitochondrial ceramide should be recognized as one of the underlying factors leading to mitochondrial dysfunction with age.
Involvement of DNA Damage in Age-Related Mitochondrial Dysfunction
It is important to recognize that mtDNA is replicated in postmitotic cells such as cardiomyocytes. An increase in mtDNA copy number in the absence of cell division has been observed in aged mouse heart as a part of mitochondrial biogenesis stimulated by oxidative damage (see previous sections). Upregulation of mtDNA copy number is concomitant with significant upregulation of the master regulator peroxisome proliferator-activated receptor-γ coactivator-1-α (PGC 1α) and its downstream transcription factor network, including mitochondrial transcription factor A (TFAM) and nuclear respiratory factors (NRFs) [13].
One well-recognized process associated with aging is an increase in frequencies of mtDNA point mutations and deletions. These frequencies increase ∼threefold in old mouse hearts, compared with young adult hearts [13]. One of the earliest studies showed that the amount of 5 kb-“common deletion” of mtDNA (mtDNA4977) in the heart and brain of older individuals was around 0.1% but was undetectable in fetal tissues [99]. Another study reported the appearance of this and two other deletions (mtDNA7436 and mtDNA10422) in the heart beginning at 40 years of age with subsequent accumulation with increasing age [100]. The highest levels of the mtDNA7436 deletion in human hearts (3–9%) are detected in 80- and 90-year-old individuals [101]. The role of mtDNA aberrations in aging has been assessed in experiments on mice with disabled DNA-proofreading property of mitochondrial polymerase γ (the mtDNA mutator mouse model). Animals with mutated mitochondrial polymerase γ demonstrate substantial increase in mtDNA point mutations and deletions [6, 7], shortened life span, and a phenotype of accelerated aging, including age-dependent cardiomyopathy [7]. Furthermore, increased levels of mtDNA mutations have been shown to accelerate apoptosis (see below). Mutations in the genes encoding the individual components of ETC may also be one of the mechanisms of age-associated destabilization of a native architecture of ETC supercomplexes recently discovered by Gomez et al. [41].
Mutated mtDNA often coexists with normal mtDNA, a condition referred to as heteroplasmy, and the levels of mutated mtDNA could vary dramatically between tissues [102, 103]. The mutated mtDNA can be accumulated to different levels in the individual cells, but it has to be present at a certain minimal threshold level to cause mitochondrial dysfunction, such as respiratory chain deficiency. Interestingly, in muscle cells with high proportions of mutated mtDNA, mitochondrial biogenesis is activated as a compensatory but ineffective response to ETC deficiency. As a result, such cells contain the massive amount of abnormal mitochondria.
In most reported cases, mutations in aging tissues are single large mtDNA deletions. Thus, accumulation of mtDNA deletions better correlates with the premature aging in mice than are mtDNA point mutations [104]. (Note: this is not the case of the mtDNA mutator mouse in which accumulated mtDNA point mutations rather than large mtDNA deletions are driving force of premature aging [105].) Age-associated accumulation of respiratory chain-deficient cells with mtDNA deletions have been documented in various human tissues, including heart [99, 106, 107]. They accumulate clonally leading to the formation of mosaic respiratory chain deficiency in aging organisms. Mosaic respiratory chain deficiency has been reported in aging human heart [108]. The frequency of respiratory chain-deficient cells is substantial and could well contribute to age-associated organ dysfunction [109].
It is often assumed that the age-associated somatic mtDNA mutations are generated due to oxidative damage and therefore start to accumulate in aging adults. This idea has been supported by a wealth of correlative data showing higher levels of somatic mtDNA mutations in older than in younger humans and other mammals. Moreover, overexpression of antioxidative enzyme (e.g., catalase) in mitochondria significantly reduces frequencies of age-related mtDNA mutations and deletions and mitochondrial biogenesis presumably via attenuation of age-dependent mitochondrial oxidative damage [13]. However, an alternative possibility has recently been suggested that somatic mtDNA mutations are generated by replication errors and that many of the somatic mtDNA mutations in adults can be traced back to embryonic development or early postnatal life. In support of this hypothesis, most of the human mtDNA mutations are generated by replication errors and not by damage [110], and oxidative damage of mtDNA in the form of guanine adduct 7,8-dihydro-8-oxo-deoxyguanosine (8-oxodG) is not a major contributor to the dysfunction of mitochondrial respiratory chain [111]. In aging, loss of apoptotic cells, related to the development of respiratory chain deficiency, is often accompanied by increased oxidative stress, although it has not been observed in all examined cases. For instance, a massive increase of apoptosis in the mice with abolished mtDNA expression and in the mtDNA mutator mice takes place without any significant oxidative stress [6, 112, 113]. Moreover, the mtDNA mutator mouse gives additional support to the hypothesis of early postnatally originated mitochondrial senescence as it contains abundant mtDNA mutations in mid-gestation, but develops symptoms and focal respiratory chain deficiency during adult life [7].
Loss of Cardiac Cells due to Chronic Exposure to Free Radicals in the Senescent Myocardium
A decline in cardiac function during the aging process involves a significant loss of cardiomyocytes (∼30%), hypertrophy of the remaining cells, and increased growth of connective tissue [114–117]. An important process mediating the loss of cardiac cells with advanced age is the programmed cell death, apoptosis [28, 118].
Ljubicic et al. [119] have demonstrated that aging evokes alterations in rat cardiac mitochondria composition and function, which are suitable for induction of mitochondrially mediated apoptosis. In particular, mitochondria isolated from hearts of aged animals exhibit lower respiratory control ratio and mitochondrial respiratory capacity, compared to mitochondria from young animals. Dysfunction of senescent mitochondria is accompanied by increased ROS production indicating that cardiac mitochondria of aged animals are subject to chronic oxidative stress.
Elevated ROS levels trigger the opening of the MPTP and facilitate the progression of the apoptotic program. MPTP is located in the MIM. When fully opened, this voltage-dependent “channel” enables passive diffusion of molecules with molecular masses up to 1.5 kDa which might cause swelling of mitochondria, drop of mitochondrial membrane potential, and depletion of ATP, with subsequent apoptosis and cell death (see Chap. 3). Cardiomyocytes from aged myocardium are more susceptible to induction of MPTP opening compared to those from young heart [73, 119, 120], and ROS are involved in this process in the aging heart [121]. It has been shown that peroxidized cardiolipin induces the MPTP opening in isolated rat heart mitochondria [122]; therefore, it is conceivable to hypothesize that increased ROS-dependent peroxidation of cardiolipin with age enhances MPTP opening (Fig. 13.2). In accordance with this hypothesis, an antioxidant melatonin, which inhibits cardiolipin peroxidation in rat heart mitochondria [123], protects aging mitochondria against MPTP opening [73]. Interestingly, in senescent hearts, interfibrillar subpopulation of cardiac mitochondria is more susceptible to MPTP opening and release of proapoptotic-inducing factors compared to subsarcolemmal organelles [124].
Increased ROS production and MPTP opening are two major pro-death factors responsible for a key role of mitochondria in regulating apoptosis in aging heart. First, ROS can trigger mitochondria to release apoptogenic proteins, such as cytochrome c and apoptosis-inducing factor (AIF), into the cytosol [28, 125]. Cytochrome c is a part of ETC and is required for proper mitochondrial ATP synthesis. However, upon an apoptotic stress, cytochrome c is released from the organelle and proceeds to activate the cytosolic assembly of the apoptosome, which in turn results in DNA fragmentation and cell death [126]. Cytosolic accumulation of cytochrome c has been shown to be greater in the hearts of old rats, indicating that the release of this apoptogenic protein is higher in response to advanced age [73, 119]. It has been shown that the binding of cytochrome c to outer surface of the MIM is cardiolipin-dependent [127]. Oxidative damage of cardiolipin leads to the detachment of cytochrome c from the MIM, an initial step in the proapoptotic release of cytochrome c from mitochondria (Fig. 13.2) [63, 128, 129]. Thus, enhanced oxidation of cardiolipin in the aging rat myocardium promotes cytochrome c release from mitochondria [122], and this process can be counteracted by treatment with antioxidant melatonin [73, 123].
Cell susceptibility to apoptosis is also influenced by the family of “B-cell lymphoma 2” (Bcl-2) proteins. They function in accordance with their intracellular localization. Expression of one of the proteins from this family, Bcl-x, involves the alternative splicing of Bcl-x gene. One splice product, Bcl-xL, is the antiapoptotic protein. In unstimulated cardiac cells, Bcl-xL predominates, while the proapoptotic splice isoform, Bcl-xS, is barely detectable. It turns out that the cardiac Bcl-x isoform plays an important role in mitochondrial quality control, which contributes to the final removal of aged cardiac cells. A crucial mechanism for the induction of mitochondrial dysfunction and mitochondria-dependent apoptosis via an alteration in the balance of Bcl-x splice isoforms is an inhibition of neuregulin/erbB-receptor signaling pathway [130, 131]. The attenuation of erbB-receptor signaling in rat cardiomyocytes leads to mitochondrial dysfunction (membrane potential reduction, decrease ATP production) and accompanied by release of cytochrome c, activation of caspase-9 and caspase-3, and cleavage of nuclear DNA leading to mitochondria-dependent apoptosis [131]. Attenuated signaling of neuregulin via erbB-receptors is observed in aging rat myocardium [132]; therefore, decrease of neuregulin/erbB signaling may be responsible for mitochondrial dysfunction and apoptotic cardiomyocyte losses in the aging myocardium.
While several pro- and antiapoptotic Bcl-2 proteins reside in the mitochondria, Bax is the cytosolic protein in healthy cells. In a proapoptotic environment, Bax translocates to the MOM [133–135] and promotes release of proapoptotic proteins and loss of mitochondrial membrane potential [134, 135]. In addition, Bax promotes fragmentation of mitochondria by interaction with the mitochondrial fission machinery [136–140]. The greater susceptibly for mitochondrial membrane permeabilization via the opening of the MPTP in mitochondria isolated from the hearts of senescent rats accelerates subcellular translocation of apoptogenic proteins both to and from the mitochondrial compartment. Thus, cardiac mitochondria isolated from senescent rats are enriched in Bax [119], indicating increased proapoptotic signaling, compared to those isolated from younger animals (Fig. 13.2).
AIF is a caspase-independent death effector normally found behind the MOM. When the mitochondria are damaged, it moves to the cytosol and finally ends up in the nucleus, where it activates condensation of chromosomes and fragmentation of DNA in order to prepare for cell death. AIF release from mitochondria to cytosol is enhanced in the aged myocardium and parallels elevated level of nuclear DNA apoptotic fragmentation [119] (Fig. 13.2).
A critical component of the apoptotic response to oxidative stress and regulator of life span in mammals is the “human Src homology and collagen” (Shc) protein of 66-kDa (p66Shc) [141–143]. Oxidative stress triggers a mitochondrial accumulation of the protein, and once imported into the organelle, p66Shc causes alterations in mitochondrial Ca2+ handling and three-dimensional structure, inducing thereby apoptosis [144]. In addition, p66Shc oxidizes cytochrome c and catalyzes H2O2 synthesis [142]. Thus, one mechanism [142] by which this protein facilitates the progression of the apoptotic program is an oxidation of cytochrome c making it unable to reduce oxygen to water. As a result, a fraction of the mitochondrial electron flow produces hydrogen peroxide, which in turn induces opening of the MPTP. An association between p66Shc and cardiomyocyte apoptosis under conditions of cardiac stress has been demonstrated in a number of studies [145–148]. Moreover, data of Ljubicic et al. [119] clearly demonstrate the accumulation of p66Shc in the mitochondrial fraction obtained from the hearts of aged rats. In contrast, mice with a targeted mutation of the gene encoding p66Shc demonstrate prolonged life span, reduced production of intracellular oxidants, and increased resistance to oxidative stress-induced apoptosis [141]. Recently, new insights into the mechanism of ROS-regulated translocation of p66Shc to mitochondria have been provided. In mouse embryonic fibroblasts, free radicals activate protein kinase Cβ isoform to phosphorylate serine-36 on the p66Shc, allowing it transfer to mitochondria [144]. Within the mitochondrion, several components of protein-importing TIM–TOM system (e.g., 70-kDa heat-shock protein, mtHSP70; translocase of the MIM, TIM44) bind and suppress activity of phospho-p66Shc. Proapoptotic stimuli destabilize this interaction leading to release of p66Shc with subsequent oxidation of cytochrome c and production of hydrogen peroxide (Fig. 13.2). Interestingly, senescent myocardium demonstrates a greater level of expression of mtHSP70 [149]. The upregulation of this protein in the hearts of aged rats may serve a compensatory adaptation—an effort to limit the availability of the apoptogenic p66Shc within the mitochondria.
There is evidence that multiple components of the mitochondrial morphogenesis machinery, including both mitochondrial fission and fusion mediators, can influence apoptosis [137, 150–155]. For instance, dynamin-related protein 1 (Drp 1), when stimulated, translocates from the cytosol to the mitochondria to participate in the release of mitochondrial proapoptotic factors [137, 152, 155] and apoptotic mitochondrial fragmentation [150, 152, 156]. Similarly, optic atrophy type 1 protein (Opa 1) is involved in the release of cytochrome c during apoptosis [151, 153, 154]. According to Ljubicic et al. [119], the overall cellular content of Drp 1 and Opa 1 is elevated in the senescent rat myocardium, suggesting a greater mitochondrial dynamism (increased Drp 1/Opa 1-regulated mitochondrial fusion/fission events) as compensatory mechanism against reduced mitochondrial content and function in aged heart.
Aging and Biogenesis of Mitochondria
Mitochondrial biogenesis decreases in the aging myocardium. Several global transcriptional nuclear regulators of gene expression, including the fatty acid peroxisome proliferator-activated receptor (PPAR) and its coactivator (PGC1), are involved in the control of mitochondrial biogenesis. Myocardial expression of PPARα and PGC1 is lower in aged rats compared to young rats [29, 157]. PPARα is present at significantly higher levels in exercise-trained aged animals compared to sedentary aged ones. Age-associated decrease in PPARα correlates with the expression of several enzymes involved in mitochondrial FAO [e.g., 3-hydroxyacyl-CoA dehydrogenase (HAD), carnitine palmitoyltransferase-I (CPTI)] because they are encoded by PPARα-regulated genes [157]. In general, age-related transcriptional downregulation has been reported for many genes involved in fatty acid uptake and oxidation (CD36, hormone-sensitive lipase, CPTs, carnitine acyltranferase, acyl-CoA dehydrogenase, etc.) (Fig. 13.1) [23].At the same time, genes involved in carbohydrate metabolism (Glut4, phosphofructokinase, enolase) are upregulated in the aged myocardium. Apparently, transcriptional alterations underlie a metabolic shift from fatty acid to carbohydrate metabolism, associated with aging heart. Interestingly, in the rodent model caloric restriction (CR) prevents the age-enhanced transcriptional changes and several age-dependent physiological and biochemical alterations [23].
Preston et al. [29] have demonstrated widespread transcriptional suppression of the mitochondrial energy production system in the aged rat ventricles. According to their data, transcription of 16 out of the 37 complex I genes encoded by nuclear DNA is decreased in old rat heart. Interestingly, no changes in any of seven mtDNA-encoded complex I subunits have been observed in this study. Similarly, 2 genes encoding subunits of complex II, 7 genes encoding subunits of complex III, 8 of the 17 nDNA-encoded complex IV genes (and none of three mtDNA-encoded subunits), and 9 of the 21 genes that encode subunits of complex V are downregulated in the aged heart. It is worth to note that broad age-associated transcriptional downregulation of mitochondrial genes is accompanied by selective decrease in activities of only OXPHOS complexes I and V (Fig. 13.1) [29].
Data on the role in aging of nuclear regulators of mtDNA transcription and replication are contradictory. While mitochondrial transcription factor (TFAM), which is implicated in both mtDNA replication and transcription, has not been changed in the aging rat heart [158, 159], nuclear respiratory factors (NRF1, NRF2), which are global regulators of mitochondrial transcription, have been upregulated [160] or downregulated [161] in the aging heart. At this point, it is not a big surprise that literature on the age-related changes in myocardial mtDNA-encoded proteins is very controversial. Ventricular cardiomyocytes from aging rodents have been shown to express decreased levels of cytochrome b, component of complex IV (COXIII) [162], and to display reduced mtDNA transcription (Fig. 13.1) [163–165]. However, in other studies no significant changes [160, 166] or increased transcription of mitochondrial genes [167] have been also reported. Similarly, inconsistent data exist in regard to the levels of mtDNA: Increased [159, 168] as well as unchanged [158, 166, 169, 170] levels have been reported in senescent heart. It is evident that further research is needed to elucidate the role that nuclear regulatory factors and nuclear-encoded enzymes play in mitochondrial biogenesis in aging heart. One potential explanation of discrepancy in findings is the existence of subpopulations of cardiac mitochondria with different age-related loss of function. Indeed, it has been shown that age-related defects such as decrease of complex III [171, 172], COX activity, and OXPHOS [173] are limited to subpopulation of interfibrillar cardiac mitochondria: these changes correlate with a marked decline in interfibrillar (but not sarcolemmal) mitochondria levels of antioxidants, ascorbate, and reduced glutathione [20].
Mitochondrial Dynamics in Aging
Important factors in several key pathologies associated with aging are functional and structural alterations in mitochondrial morphology. The nature of mitochondrial morphology is very dynamic and is determined by the balance between mitochondrial fusion and fission. Mitochondrial fusion and fission proteins are also implicated in mitochondrial metabolism, redox signaling, maintenance of mitochondrial DNA, and removal of damaged mitochondria from the cell [174]. Impairment of mitochondrial dynamics might be one of the intrinsic causes of mitochondrial dysfunction, which contributes to oxidative stress and cell death during the aging process.
It has recently been shown that in the skeletal muscle of aging individuals, the expression of the mitochondrial fusion mediator protein, mitofusin 2 (Mfn2), and regulator of mitochondrial fission, dynamin-related protein 1 (Drp1), are reduced [175]. Decrease of these important mitochondrion-shaping proteins seems very likely to cause an imbalance between mitochondrial fusion and fission with subsequent impairment of mitochondrial function and age-related loss of muscle. Furthermore, it has been found that expression of Mfn2 is regulated by PGC1α [176, 177]. The level of PGC1 decreases with age (see above) suggesting the existence of a pathway that contributes to aging of mitochondria via changes in PGC1α-mediated regulation of fusion/fission-specific genes.
Telomeres
It has been shown that cardiomyocyte renewal rate decreases two times in elderly human hearts compared to young adults [178]. This phenomenon could be explained by the decline in number and regenerative capacity of cardiac stem cells. For example, Anversa et al. [179] have reported that cardiac stem cells in older animals and patients with cardiovascular diseases have a higher rate of apoptosis, shorter telomeres, and increased expression of the senescence marker p16INK4a.
Replicative capacity of cells highly depends on the length of telomeres, the ends of chromosomes. Shortening of telomeres serves as a trigger of replicative senescence in normal cells [180], and overexpression of the catalytic subunit of telomerase, an enzyme elongating telomeres, leads to cell immortalization [181].
There is growing evidence supporting an involvement of mitochondria in replicative senescence of cells. Quite a large number of independent studies have shown telomere shortening as a stress response. Exposure to mild oxidative stress leads to faster telomere shortening, whereas maintenance of telomere length improves under low oxidative stress [182]. As it has already been discussed, mitochondrial function declines and mitochondria-originated oxidative stress increases with age. Thus, elevated ROS generation could limit cell proliferation through telomere shortening; therefore, telomeres can be viewed as sensors of damage in the mitochondria [183]. In support of this hypothesis, selective targeting of antioxidants to the mitochondria lessens telomere shortening and increases life span in fibroblasts under mild oxidative stress [184], while experimentally induced mitochondrial dysfunction leads to an increase in ROS production of and telomere loss in mouse embryos [185].
Cardiac Channelopathies
One of the consequences of aging-associated detrimental changes in mitochondrial proteins, lipids, and mtDNA discussed earlier is the malfunctioning of mitochondrial ion channels. The relationship between the activity of multiple ion channels found in mitochondria and cellular aging has been intensively studied. Defects in mitochondrial ion channels, often leading to their pathological dysfunctions, mitochondrial channelopathies, are found in a large spectrum of human pathologies including aging. Channelopathies may be the result of mutations in genes encoding ion channel proteins, which disrupt channel function or change their expression, or mutations in channel-regulating proteins leading to changes in the activity of non-mutated channels. Moreover, harmful posttranslational modifications of proteins participating in ion-channeling may also result in channelopathies. In this section we describe several mitochondrial ion channels whose dysfunction is associated with cardiac aging or may affect the aging processes: Ca2+ transporters, mitochondrial ATP-sensitive potassium channel (mitoKATP), voltage-dependent anion channel (VDAC), as well as MPTP (Fig. 13.3).
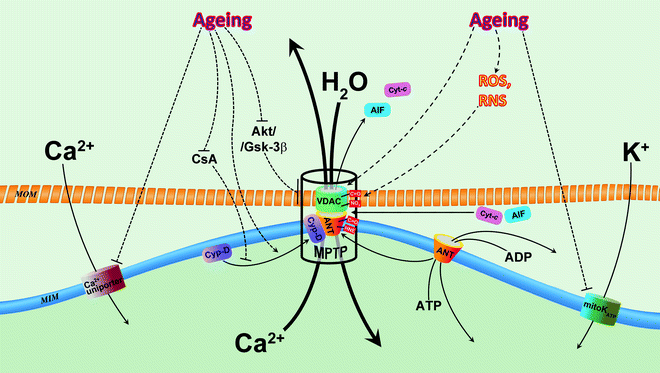
Fig. 13.3
Alterations of mitochondrial ion channels in aging. Two mitochondrial channels contribute to age-associated decrease of mitochondrial Ca2+: Ca2+ uniporter and mitochondrial permeability transition pore (MPTP). Age-associated inhibition of Ca2+ uniporter decreases entering of Ca2+, whereas activated MPTP facilitates extrusion of Ca2+ from mitochondrial matrix. Decreased concentration of Ca2+ dampens its stimulatory effects on bioenergetic metabolism in senescent myocardium via reduction of TCA activity, OXPHOS and ATP synthesis. It has been suggested that mitochondrial ATP-sensitive K+-selective channel (mitoKATP) is impacted in aging, and this results in reduced cardiac tolerance to stress and loss of preconditioning cytoprotection. Oxidative stress increases with age and leads to overproduction of reactive oxygen species (ROS) and reactive nitrogen species (RNS). ROS and RNS directly, via carbonylation (C=O) and nitration (NO2), and indirectly, via formation of lipid peroxidation product 4-hydroxynonenal (HNE), modify major components of MPTP, voltage-dependent anion channel (VDAC), adenine nucleotide translocator (ANT), and cyclophilin-D (CyP-D) which results in the sensitization/activation of this superchannel with subsequent loss of mitochondrial Ca2+, collapse of mitochondrial membrane potential (ΔΨm), mitochondrial swelling, and release of apoptogenic factors, leading to cell death. Moreover, dysfunction of MPTP regulation in the senescent myocardium (see text for details) may contribute to loss of cardioprotection with aging. Abbreviations: AIF apoptosis-inducing factor, Akt protein kinase B, CsA cyclosporine A, Cyt-c cytochrome c, Gsk-3β glycogen synthase kinase 3 beta, MIM mitochondrial inner membrane, MOM mitochondrial outer membrane. Solid arrows indicate movements/translocations of molecules. Dashed arrows denote regulatory influences (inhibition/stimulation of function, expression, modification of proteins, etc.)
Ca2+ plays an important role in the activation of mitochondrial Ca2+-dependent dehydrogenases, which supply reducing equivalents for OXPHOS and ATP synthesis. Jahangir et al. [186] have demonstrated that mitochondrial Ca2+ content in the heart from the senescent rat was significantly reduced compared to adult rat. This finding is consistent with the observation that activity of Ca2+-dependent NAD isocitrate dehydrogenase (NAD-IDH) is decreased in aged rat heart [187]. Therefore, reduced efficiency of ATP synthesis in aging mitochondria may not only associate with decreased ETC enzyme activities but also with reduced mitochondrial matrix Ca2+ levels and with depressed Ca2+-dependent dehydrogenase activities. The reduction in Ca2+ content could result from decreased Ca2+ uptake in aging mitochondria—the process driven by mitochondrial ΔΨm-dependent ruthenium-red-sensitive Ca2+ uniporter. Senescent cardiac mitochondria display both a 15% reduction in ΔΨm and a slower rate of mitochondrial Ca2+ uptake (approximately 20%) compared to adult mitochondria [186]. Thus, it seems likely that decreased ΔΨm is responsible for reduced mitochondrial Ca2+ uptake (Fig. 13.3). The molecular identity of this channel has recently been determined [188], which allows to clarify whether changes on the expression of this channel contributes to aging-mediated channelopathy.
MPTP when opened is permeable for Ca2+. Activation of this pore is enhanced in the mitochondria of senescent cardiomyocytes [189], (see below). MPTP opening is more susceptible to activation in heart mitochondria from senescent Fisher 344 rats compared to mitochondria from adult heart [186], suggesting that reduced Ca2+ content in mitochondria with aging may be also the result of elevated Ca2+ extrusion through MPTP (Fig. 13.3). Mitochondrial Na+/Ca2+ exchanger (NCX) is another major extruder of Ca2+ from mitochondria, but its role in disturbed Ca2+ handling in aging is undefined.
The MIM contains K+-selective channel, mitoKATP, which belongs to the inward rectifier K+ channel family. This channel is blocked by ATP and a number of inhibitors and is activated by K+ channel openers and superoxide anion. The molecular identity of mitoKATP is not yet known. mitoKATP activation in the heart is generally considered as a contributory effector component in the preconditioning pathway (and may be involved also in the triggering of the ischemic preconditioning response) [190]. Aged rat hearts subjected to ischemia-reperfusion have exhibited more severe tissue and function damage than adult, suggesting an increased susceptibility to damage in elderly hearts (reduced myocardial tolerance to ischemia-reperfusion injury) [191]. Moreover, several studies have suggested that the preconditioning mechanism is insufficient or lost in aged or senescent hearts [192–194]. Based on these observations, it has been suggested (albeit indirectly) that mitoKATP channels are involved in aging: mitoKATP channel defects may, in part, lead to ineffective preconditioning in aging hearts (Fig. 13.3).
The function of one of the mitochondrial channels—MPTP—is quite paradoxical. When activated/opened, it is nonspecifically permeable to solutes of molecular mass up to ∼1.5 kDa, so solutes with molecular masses up to about 1.5 kDa cross the mitochondrial inner and outer membranes, whereas matrix proteins are not permitted to diffuse through the pore. Therefore, the opening of MPTP leads to the osmotic force of matrix proteins, and the accumulation of water and matrix volume increase (swelling). As a consequence of MPTP opening, cell death is triggered by the collapse of ΔΨm, ATP hydrolysis, MOM rupture, and the release of proapoptotic factors (cytochrome c, AIF.). Given this damaging potential of MPTP, it is closed under physiological conditions.
The MPTP undergoes conformational change and forms an active nonselective channel under stress conditions such as ischemia-reperfusion and Ca2+ overload. This results in the above-mentioned harmful changes in mitochondria and eventually to cell apoptosis. An increase of MPTP opening is also age-associated, as it was first demonstrated in mice liver [195]. Later an increased susceptibility to MPTP opening has been found in cardiac mitochondria of old rats: aging animals demonstrate reduced mitochondrial Ca2+ retention capacity [186] and greater mitochondrial swelling (Marin-Garcia et al., unpublished data) than young adults.
Although the mechanism of the MPTP functional changes during aging is unknown, it has been suggested that oxidative stress plays an important role in this process. Oxidative stress increases with age in cardiac mitochondria in parallel with reduction of endogenous antioxidant agent, reduced glutathione [21]. As a result, elevated ROS and RNS may oxidatively modify and activate components of MPTP. For instance, voltage-dependent anion channel (VDAC), an important subunit of MPTP located in the MIM, undergoes age-dependent nitration in rat myocardium [196]. Although it is yet not known whether this oxidative modification alters functional properties of VDAC, there is evidence that another oxidative radical, O2•−, induces VDAC-dependent cytochrome c release and enhances sensitivity of MPTP to Ca2+ [197]. Similarly, ROS in aging-related manner carbonylate redox-sensitive cysteine residues on adenine nucleotide translocator (ANT), another essential component of MPTP, located in the MIM [198]. Carbonyl modification reduces capability of ANT to exchange ADP/ATP across the MIM with subsequent uncoupling of OXPHOS and collapse of ΔΨm. Moreover, oxidation may switch ANT to a nonselective pore, permeable for small ions and metabolites (Fig. 13.3). Furthermore, ANT is known to be regulated by 17.6/18.6-kDa proteins, cyclophilin-D (CyP-D): Binding of CyP-D to ANT in the presence of high mitochondrial Ca2+ converts ANT from adenine nucleotide transporter to MPTP channel component and sensitizes the latter to Ca2+ [199]. CyP-D contains relatively polyunsaturated fatty acids that could be oxidized by ROS to produce lipid peroxidation products, HNE and 4-hydroxyhexenal (HHE). They may modify ANT to inhibit its ADP/ATP-transporting activity and to facilitate interaction with CyP-D [200, 201]. As we have mentioned, this makes an ANT a component of active MPTP and sensitizes MPTP to Ca2+. Besides oxidative stress, changes in the expression of major MPTP components seem to play a role in the increase of the permeability of mitochondrial membranes in aging. Thus, according to Chorna et al. [202], expression of VDAC and ANT increases 1.7–1.8 times in the old rats’ hearts in comparison with adult animals (Fig. 13.3).
Opening of MPTP following ischemia-reperfusion injury can be prevented by cyclosporine A (CsA). This immunosuppressant binds to CyP-D and prevents the binding of Cyp-D to the ANT [203]. Recently, Liu et al. [204] have reported that in old rat cardiomyocytes, CsA fails to reduce MPTP opening. Interestingly, negative regulation of MPTP via Akt/GSK-3β signaling pathway also is compromised in the old myocardium. Thus, isoflurane-dependent phosphorylation of Akt and GSK-3β correlates with its cardioprotective effect (inhibition of MPTP in ischemia/reperfusion injured myocardium) in young rats, whereas anesthetic fails to prevent MPTP opening induced by ischemia/reperfusion injury in old animals [205] (Fig. 13.3). These findings suggest that MPTP regulation in the senescent myocardium is dysfunctional and could be the reason for loss of cardioprotection with aging.
In conclusion, deciphering the molecular mechanisms of age-related changes in mitochondrial channels is of great significance in terms of designing therapeutic agents to improve mitochondrial function and slowing myocardium aging.
Conclusions
Cardiac aging in human and mice includes cardiac hypertrophy, fibrosis, diastolic dysfunction, reduced functional reserve, and adaptive capacity to stress. These changes increase the risk of heart failure. Mitochondria, which are particularly active in myocardial cells, are the main source of ROS formation, which appears to play an important role in cardiac aging. Same time, mitochondria are the main target for free-radical attack. Aging results in nonuniform decline in activities of ETC complexes, which may be due, at least in part, to mitochondrial oxidative stress. Overall effect of increased oxidative modification of the ETC components may be an underlying mechanism to the development of age-associated state of chronic stress and cause increased mitochondrial dysfunction leading eventually to cardiac dysfunction. Furthermore, several mitochondrial ion channels undergo structural and functional alterations with aging. These changes contribute to the development of age-associated mitochondrial dysfunction and cellular senescence in the form of age-associated channelopathy.
Among the numerous constitutive changes that occur in cardiac cells with increased age, there are a distinct decrease in the ratio of mitochondrial membrane ω-3 to ω-6 PUFA and a decrease in the mitochondrial phospholipid, cardiolipin. The age-associated increase in ω-6 PUFA, the preferred target of ROS-induced peroxidation, may be one of the mechanisms responsible for increased oxidative modification of key mitochondrial proteins in the aging heart.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


