(1)
The Molecular Cardiology and Neuromuscular Institute, Highland Park, NJ, USA
Abstract
Ventricular dysrhythmias represent the most predominant etiology of sudden cardiac death throughout the world. However, the mechanisms underlying fatal ventricular dysrhythmias are not fully understood. Over the last two decades, a growing body of evidence supports the concept that cardiac mitochondria may be involved at least in part in the genesis of cardiac dysrhythmia. The principal objective of this chapter is to describe the role that mitochondria play in altering the heart’s electrical function by introducing heterogeneity into the cardiac action potential. A focus will be placed on how the energetic status of the mitochondrial network can alter sarcolemmal potassium fluxes through ATP-sensitive potassium channels, thereby creating a “metabolic sink” for depolarizing wave fronts and introducing conditions that favor generation of fatal dysrhythmia. Mechanisms by which mitochondria depolarize under conditions of oxidative stress are characterized, and the contributions of several mitochondrial ion channels to mitochondrial depolarization are reviewed. The inner membrane anion channel in particular opens upstream of other inner membrane channels during metabolic stress and may constitute an effective target to prevent the metabolic oscillations that enhance action potential lability. Finally, therapeutic strategies that may prevent dysrhythmias by preserving mitochondrial membrane potential in the face of oxidative stress will be discussed, supporting a concept that treatments aimed at cardiac mitochondria may have the potential in attenuating electrical dysfunction in the heart.
Introduction
Among the different manifestations of cardiovascular disease, a significant cause of mortality is sudden cardiac death resulting from malignant ventricular dysrhythmia. Globally, the frequency of sudden cardiac death is remarkably similar in different regions, affecting approximately 1 of every 1,000 people and accounting for as much as one-third of all cardiac deaths in higher risk populations [1]. Cardiac dysrhythmia is a common and sometimes lethal manifestation of many acquired and inherited diseases affecting the cardiovascular system. Novel treatment aimed to reduce the incidence of sudden cardiac death clearly may have enormous potential for global health. Investigations into the electrical function of the heart began over 150 years ago when Kolliker and Muller demonstrated that the heart produced electricity accompanied by muscle contraction [2]. In 1883, Sydney Ringer first described the ionic basis for heart function [3]. Beginning in 1949 with the first reports of cardiac action potential recordings [4], including normal [5] and diseased human heart [6], major strides have been achieved in understanding mechanisms of normal and abnormal cell electrophysiology and heart rhythm generation. In the 1960s, compounds such as lidocaine was first used to treat dysrhythmia by inhibiting sarcolemmal ion fluxes, and imaging techniques with increasing resolution are constantly improving the insight into tissue-level events that lead to dysrhythmia. Despite these technical advances in understanding and diagnosing cardiac rhythm disturbances, the underlying mechanistic bases for cardiac dysrhythmias still await full elucidation, reflecting a window for therapeutic potential as these subcellular pathways responsible for aberrant conduction are illuminated.
As a syncytium, coordinated electrical propagation throughout the heart is essential for adequate contractile function. At the cellular level, each individual myocyte must depolarize and repolarize in a specific manner based on anatomical location. Pathological heterogeneity in the cardiac action potential is commonly linked to ventricular dysrhythmias, and several subcellular factors can contribute to lability in action potential duration. Among the cellular elements implicated in the induction of cardiac dysrhythmias, ion channels in the sarcolemmal and mitochondrial inner membranes have received considerable attention for their ability to influence action potential duration [4]. At the cellular and tissue levels, ion channel current is the sum of biophysical biochemical (phosphorylation and dephosphorylation), and biogenic (biosynthesis, processing, trafficking, and degradation) properties. Sarcolemmal ion channel mutations leading to prolongation of the action potential, (e.g., in the long QT syndrome), early or delayed after depolarizations due to activation of calcium channels/exchangers, and altered transsarcolemmal ion gradients have all been extensively researched for their dysrhythmogenic role. In this chapter, the role that cardiac mitochondria may play in influencing cardiomyocyte action potential duration, underscoring therapeutic potential for dysrhythmia using mitochondria-targeted approaches, will be reviewed. A focus will also be placed on the etiology of ventricular dysrhythmias evoked under conditions of oxidative stress, highlighting potential preventative approaches taken from the animal literature, with pertinent references from clinical studies where available.
The Role of Sarcolemmal KATP Channels in Dysrhythmia
Numerous studies have supported the concept that the mitochondria induce nonphysiological spatiotemporal heterogeneity in the cardiac action potential and predispose the heart to reentrant dysrhythmia. The influence of mitochondrial energetic status on the sarcolemmal action potential is mediated in large part by energy-sensing ATP-sensitive potassium channels (sarcKATP) in the sarcolemmal membrane. A significant amount of attention has been devoted to the role that sarcKATP channels may play in inducing action potential heterogeneity, leading to cardiac dysrhythmias [7, 8]. First discovered by Noma and coworkers in the early 1980s [9], myocardial sarcKATP channels are heteromultimers composed of four pore-forming subunits and four accessory subunits, the sulfonylurea receptors, that bind to ATP. Inhibited by intracellular ATP and activated by ADP, Pi, Mg, and/or pH, sarcKATP channels open in the face of oxidative stress to produce an inwardly rectifying background current, usually observed within the first 10 min of ischemia [10]. SarcKATP channels are among the most densely populated ion channels in cardiac myocardium, and the opening of even 1% of the total amount of channels in the sarcolemma can significantly shorten the cardiac action potential [11].
The opening of sarcKATP channels may be an endogenous protective mechanism of the myocardial tissue, where channel opening signaled by inadequate ATP supply decreases calcium-mediated cardiac energy demand. As the population of sarcKATP opens, the cardiac action potential shortens and reduces the calcium transient. Since calcium overload can lead to necrotic and apoptotic cell death, sarcKATP channel opening is believed to be cytoprotective by decreasing the extent contracture by the myofilaments and blunting mitochondrial calcium overload. Several lines of evidence indicate that the expression of functional sarcKATP channels is vital to cellular survival in the face of oxidative stress. First, increased sarcKATP protein expression is correlated with protection against ischemia/reperfusion injury in female (vs. male) animals [12–14] or following exercise training [12, 15]. Second, pharmacological blockade of the sarcKATP channel population increased cell death in hearts exposed to ischemia/reperfusion [13, 15, 16], with the block during ischemia being the critical period leading to increased injury [13]. Third, gene knockout of the sarcKATP channel pore-forming subunit produced animals that were severely intolerant to exercise and displayed enhanced sensitivity to calcium overload [17–19]. Taken all together, it appears that there is a physiological role for sarcKATP opening in attenuating cell death during ischemia. Supporting this concept are observations in humans where diabetic patients taking oral sulfonylureas to control type II diabetes were at a higher disposition for cardiac injury following ischemia [20].
While the opening of sarcKATP channels appears to be protective of the viability of ischemic cardiac myocytes, the consequence of increasing potassium conductance to the whole organ predisposes to electrical dysfunction and in some cases the generation of fatal dysrhythmia [7, 21–23]. With such a high density of channels in the sarcolemmal membrane, the opening of sarcKATP channels can significantly shorten the action potential and, if enough channels open, can render cells inexcitable by holding the membrane potential very close to potassium’s Nernst equilibrium potential. This creates a significant current sink for the propagating depolarization wave, and dysrhythmias may be favored when there are local regions where the open probability of sarcKATP channels is high, i.e., where the energetic status of the cell has been compromised, a phenomenon that has been termed “metabolic sinks” [24, 25]. Presence of metabolic sinks potentially enhances propensity for dysrhythmia by influencing the effective refractory period (ERP) of the myocardium, resulting in a shortened excitation wavelength. Pathological heterogeneity in action potential duration increases the dispersion of refractoriness within the tissue and is known to promote reentry [26]. SarcKATP opening abbreviates the action potential duration and shortens ERP. SarcKATP channel openers [27] and blockers [28] decrease and increase ERP, respectively. ERP is also prolonged after knockout of sarcKATP pore-forming subunit suggesting that these features are intrinsic to the myocardium and that KATP channels in the myocardium may have an important role in protecting the heart from lethal dysrhythmias and adaptation to stress situations [29]. However, other factors may come into play during ischemia which alters the relationship between action potential duration and ERP. For example, although ischemia activates sarcKATP and shortens the action potential, a prolonged ERP may occur due to postrepolarization refractoriness [30], presumably owing to alterations in Na channel availability.
A dysrhythmogenic role for sarcKATP has been confirmed in studies using either glibenclamide which blocks both the mitochondrial and sarcolemmal isoforms of the KATP channels, or the sarcolemmal-specific HMR 1833 compounds, or HMR 1098, the sodium salt of HMR 1883. Blocking sarcKATP channels with HMR1883 decreased the incidence of ventricular dysrhythmia in rat, pig, and dog [22, 31, 32]. These findings from animal models are confirmed in clinical studies where sarcKATP channel blockers have been shown to reduce the incidence of ventricular fibrillation in humans [33, 34]. While it is conceivable that the prevention of dysrhythmias with sarcKATP blockers is due to direct inhibition of sarcKATP currents, sarcKATP blockers could theoretically also prevent dysrhythmias by indirect mechanisms. Specifically, by inhibiting sarcKATP currents and preventing action potential shortening, the ensuing cellular calcium overload may promote gap junction closure, thereby blocking reentrant wave fronts via cellular uncoupling [35].
Metabolic Alterations
The cardiac mitochondrial network produces over 95% cellular ATP and accounts for 20–30% of myocardial volume in many species ranging from mouse to man [36]. According to classic chemiosmotic theory [37], mitochondria create a proton motive force by pumping protons out of the mitochondrial matrix and then use this proton electrochemical gradient to liberate the energy needed to phosphorylate ADP to ATP by the F1Fo-ATPase. The majority of the proton motive force is comprised of the mitochondrial membrane potential (Δψm), with the magnitude of Δψm being around150 mV in energized mitochondria. Decreases in Δψm diminish the amount of free energy available to generate ATP, with mitochondria shifting to ATP hydrolysis under pathophysiological conditions when the Δψm collapses substantially (Fig. 19.1).
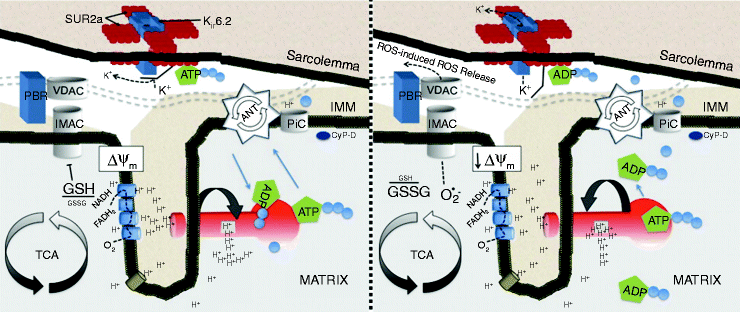
Fig. 19.1
Mitochondrial inner membrane under conditions of normoxia associated with sinus rhythm (left) and during metabolic stress (right). Cascade of events where the opening of energy-dissipating anion channels in the mitochondrial inner membrane (IMM) leads to a depolarization of the mitochondrial network, opening of sarcKATP channels, and ultimately transition to arrhythmia in the intact organ. Schematic depiction of the IMM under conditions of normoxia associated with sinus rhythm (left) and during metabolic stress (right). Matrix oxidation is characterized by glutathione oxidation and opening of IMAC, which collapses the ΔΨm, leading to ROS-induced ROS release in the mitochondrial network (adapted from Brown and O’Rourke [39] with permission from Elsevier)
The dynamic relationship between KATP current and the metabolic status of heart cells was first reported by O’Rourke and coworkers [38]. Inducing metabolic stress via substrate deprivation or in response to increased ADP levels, glibenclamide-sensitive current oscillations were observed in cardiomyocytes. Oscillating sarcKATP currents were observed in phase with NADH fluctuations and were not influenced by changing cytosolic calcium concentrations. The vacillating sarcKATP currents directly influenced cardiac repolarization and introduced significant lability in the length of the action potential waveform [38]. The energy-driven oscillations in potassium currents produced cyclical changes in the cardiac action potential and thus may contribute to the genesis of dysrhythmias during metabolic compromise. Subsequent studies confirmed the initial observation of oscillatory sarcKATP currents and action potential duration in cardiac myocytes under conditions of metabolic stress [39, 40]. The fluctuations in sarcKATP currents and consequently action potential duration are linked to the behavior of the mitochondria. Collapses in Δψm have been observed in a few studies where the myocardium is subjected to oxidative stress and with sarcKATP current increasing in phase with losses of Δψm [39].
Using cationic lipophilic rhodamine fluorescent probes, several studies have noted reversible collapses in Δψm in isolated cells subjected to oxidative stress via substrate deprivation [39, 41], ATP depletion [40], generation of reactive oxygen species (ROS) [39], the thiol oxidant diamide [42], as well as inhibition of respiration [40].
Recent evidence using two-photon microscopy has confirmed cellular data as reversible collapses in Δψm were seen in intact hearts exposed to global ischemia/reperfusion or diamide [43]. In addition to nucleotide-dependent activation of sarcKATP currents after loss of Δψm, the collapse of bioenergetics might also activate sarcKATP currents through mechanical stretch. In this scenario, the loss of mitochondrial function would then quickly preclude development of tension and result in paradoxical segment lengthening of the ischemic ventricular tissue. Given that both ischemia and stretch activate sarcKATP channels [44, 45], bulging of the myocardium may also contribute to the activation of sarcKATP channels.
Permeability Transitional Pore
The mitochondrial permeability transition pore (MPTP) is a voltage-dependent, high-conductance channel located in the mitochondrial membranes. More attention has been devoted to the activity of the MPTP in ischemia/reperfusion injury than any other mitochondrial inner membrane protein complex. Detailed discussion of the putative composition and importance of the MPTP in ischemia/reperfusion injury has been reviewed in chapters 3 and chapters 16 as well as several reviews [46, 47]. It is clear that the opening of the MPTP plays a significant role in the generation of cell death, necrotic and apoptotic, both of which are involved in pathogenesis of myocardial infarction [48]. Administration of blockers of MPTP, cyclosporin A sanglifehrin A, attenuates several indices of cardiac ischemic/reperfusion injury including myocardial infarction [49–51], left ventricular dysfunction [52, 53], cardiomyocyte death [54, 55], and mitochondrial dysfunction [56]. In newborn piglets undergoing cardiopulmonary bypass, cardioplegic arrest, weaning from bypass and reperfusion, a clinically relevant model of neonatal cardiac surgery, pretreatment with cyclosporine A prevents postcardioplegia apoptosis-related mitochondrial dysfunction [57]. The translation of these studies to clinical relevance was recently supported by a human study [58]. In 58 patients who with acute ST-elevation myocardial infarction randomized to receive either an intravenous bolus of 2.5 mg of cyclosporine per kilogram of body weight (cyclosporine group) or normal saline (control group) immediately before undergoing primary percutaneous coronary intervention, administration of cyclosporine at the time of reperfusion was associated with a smaller infarct assessed by some but not all measures than that seen with placebo.
While the role of MPTP opening in tissue death has been studied extensively, there is much less evidence that the activity of the MPTP directly influences the generation of cardiac dysrhythmia, especially those occurring at the onset of reperfusion. In several experiments using isolated cells, collapses in Δψm observed after substrate deprivation or laser flash were not prevented by the addition of cyclosporin A [39, 41, 59]. Using two-photon imaging, blocking the MPTP was ineffective at preventing the sustained Δψm collapse in hearts subjected to global ischemia [60]. Other investigators, however, reported a lack of protection against dysrhythmia in rat [61], guinea pig [24], and rabbit hearts [62]. Finally, delivery of a cyclosporin A bolus prior to stenting did not influence the incidence of ventricular fibrillation in human subjects [58]. The role of MPTP in the pathogenesis of cardiac dysrhythmias therefore remains unclear.
The Inner Membrane Anion Channel
Several distinct energy-dissipating ion channels in the inner membrane have been proposed to be involved in the Δψm collapse, thereby contributing to the generation of dysrhythmia. The first of these channels is the inner membrane anion channel (IMAC).
Anion flux across the inner mitochondrial membrane was first observed 50 years ago [63]. Earlier studies have focused primarily on the contribution of anion movement on mitochondrial volume regulation. Since the first observations, the IMAC has been characterized in several biologic tissues and is believed to play an important role in anion efflux from energized mitochondria [64]. Although the exact structure of the IMAC is not known, the sensitivity of the anion channel to regulation by benzodiazepine compounds [65] suggests that the molecular composition consists of an anion channel subunit that associates with a peripheral benzodiazepine receptor in the outer membrane.
Insights into the factors mediating the collapse in Δψm have focused on the production of ROS by the mitochondria. ROS-dependent oscillations in Δψm were first noted by Sollott and coworkers [66]. These investigators noted that local generation of ROS produced by laser flash elicited synchronous collapses in Δψm that were prevented by a ROS scavenger. There is growing evidence that the collapse in Δψm may be mediated by superoxide anion, leading to depolarizations in the myocardium through a process termed “ROS-induced ROS release” [66, 67]. According to this theory, ROS produced at the level of a single mitochondrion can stimulate superoxide-mediated depolarization of neighboring mitochondria. This spatiotemporal behavior among the mitochondrial network led others to conclude that mitochondria are arranged in a percolation matrix. According to empirical data, the increase in ROS under conditions of oxidative stress can reach a critical level, after which cell-wide Δψm oscillations in the mitochondrial network are observed [68].
The importance of IMAC in influencing the Δψm was first noted when several distinct ligands to IMAC were found to prevent loss of Δψm observed in isolated cardiac myocytes. Aon et al. used a laser flash to induce a local burst of mitochondrial ROS, which causes cell-wide increases in ROS production and oscillations in Δψm [39]. The reversible collapses in Δψm, and the cell-wide ROS accumulation, could be prevented with the addition of PK11195, 4′-chlorodiazepam, or DIDS, three distinct compounds that have previously been shown to block the activity of IMAC [69]. Furthermore, blocking the reversible collapses in Δψm by targeting the IMAC terminated the oscillations in action potential duration [39], providing further cellular evidence that targeting the IMAC may be effective in preventing dysrhythmias by stopping ROS-induced ROS release.
A series of studies whereby inhibiting the IMAC prevented dysrhythmias in intact mammalian hearts further confirms the involvement of IMAC [24, 62]. To examine whether diamide, a glutathione oxidant, caused mitochondrial depolarization and promoted dysrhythmias in normoxic isolated perfused guinea pig hearts and to investigate whether stabilization of Δψm with a ligand of the mitochondrial benzodiazepine receptor (4′-chlorodiazepam; 4-ClDzp) prevented the formation of metabolic sinks and consequently precluded dysrhythmias, oxidation of the glutathione (GSH) pool was initiated by treatment with 200 μM diamide for 35 min followed by washout. This treatment increased oxidized (GSSG) and decreased both total GSH and the GSH/GSSG ratio. All hearts receiving diamide transitioned from sinus rhythm into ventricular tachycardia during the diamide exposure. These dysrhythmias and impaired LV function were significantly inhibited by coadministration of 4-ClDzp (Fig. 19.2) [70].
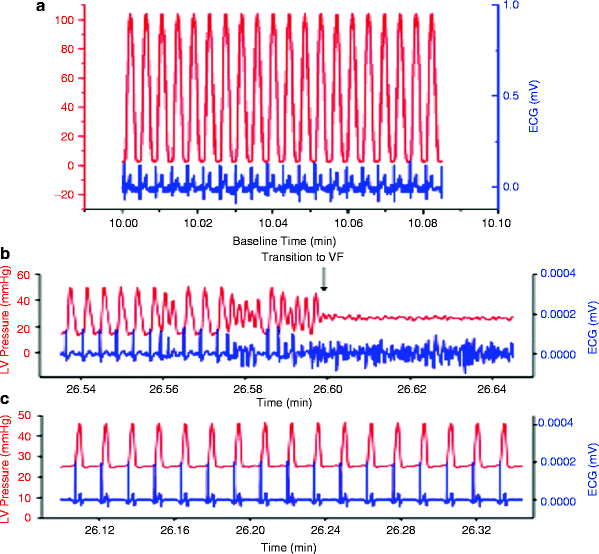
Fig. 19.2
Left ventricular pressure and ECG from hearts with diamide-induced dysrhythmias. (a) Control heart after 10 min of baseline perfusion with simultaneous LV pressure (red) and ECG (blue). (b) LV pressure and ECG in a heart 6 min into the washout period following the diamide treatment showing the transition to VF (blue) with concomitant loss of pump function (red). (c) LV pressure and ECG in a heart 6 min into the washout period following diamide treatment plus 64 μM 4-ClDzp (adapted from Brown et al. [70] with permission from Elsevier)
Optical mapping of the epicardial surface of guinea pig hearts revealed that blocking IMAC decreased ischemia-induced action potential shortening and was accompanied by a lack of a development of ventricular dysrhythmias at the onset of reperfusion [24]. Cardioprotection evoked by blocking the IMAC was also observed in isolated rabbit heart and was accompanied by significantly improved left ventricular developed pressure 74. In both studies the reperfusion dysrhythmias were prevented when the IMAC was blocked only at the onset of reperfusion as opposed to as a pretreatment [24, 62].
The Mitochondrial KATP Channels
Evidence for a mitochondrial ATP-sensitive potassium (mitoKATP) channel was first observed in the rat liver mitochondria [61, 71] and later seen in the heart as well [72]. The opening of mitoKATP channels may mediate the protective interventions administered before the onset of ischemia by partially dissipating the Δψm, reducing the driving force for calcium into the mitochondria, and while cellular respiration secondary to mild swelling of the matrix [8, 73, 74].
Most studies that have examined the potential cardioprotective effect of mitoKATP opening have focused on examining the role of mitoKATP in reducing infarct size elicited by a single preconditioning stimulus [73, 74]. In most but not necessarily all of these studies, blocking the mitoKATP with 5-hydroxydecanoate (5-HD) abolished the benefit of reduction in infarct size. While single episodes of preconditioning may provide mechanistic insight, when repetitive stimuli are administered, mitoKATP blockade does not reduce the evoked protection, as evidenced by the lack of effect of 5-HD in abolishing the infarct-reducing effects of repetitive ischemic preconditioning [75] or of chronic exercise [15]. On the other hand, few studies have examined the activity of mitoKATP channels in cardiac dysrhythmia. A role for mitoKATP in protecting against dysrhythmia is apparent where mitoKATP blockers abolished the antidysrhythmic phenotype provided by a preconditioning stimulus such as ischemic preconditioning [76], adenosine [77], δ-opioid agonists [78], estrogen [79], 3-nitropropionic acid [80], nitroglycerin [81], norepinephrine [82], or the endothelin receptor agonists. Although mitoKATP channels appear to be important in mediating the antiarrhythmic effects in some preconditioning models, their activity cannot be attributed to all models of preconditioning. For instance, blocking the mitoKATP during preconditioning from bradykinin [83], low-flow ischemia [83], peroxynitrite [84], or estradiol [85] failed to attenuate the antiarrhythmic protection. Protection against dysrhythmias via direct activation of mitoKATP channels prior to ischemia has also yielded opposing results, with some investigators showing protection from dysrhythmia [77, 86] while others showing no beneficial effect at all [75]. One putative explanation for the discordant findings is that the pharmacological agents used to open mitoKATP were different between these studies (minoxidil, diazoxide, and/or BMS-191095), and some of these compounds may be limited by their nonspecific actions.
While literature on preconditioning provides useful mechanistic insight regarding antidysrhythmic strategies administered before index ischemia, the clinical relevance of these strategies remain to be determined. To the clinician, attenuation of dysrhythmias must often be attempted after the onset of ischemia. Targeting the mitoKATP channels after the onset of metabolic stress seemed promising based on cellular experiments, where administration of mitoKATP terminated oscillations in Δψm evoked by halting respiration [40], and mitoKATP opening with diazoxide improved cellular survival and mitochondrial integrity during cellular reoxygenation [87]. Despite these encouraging cellular data, postischemic administration of mitoKATP openers does not decrease dysrhythmias [86], and effects of postconditioning interventions have been shown to be independent of the activity of mitoKATP channels [61]. Indeed, the same investigators that observed beneficial effects of diazoxide on isolated cells [87] found that the cytoprotective properties of the drug were independent of mitochondrial potassium flux [88]. The nonspecificity of commonly used mitoKATP openers, such as diazoxide and blockers, such as 5-HD is well known and has been reviewed previously [15, 74, 89, 90].
The most recent study of mitoKATP channel inhibition has involved the ischemia-exercised heart [91]. Male Sprague-Dawley rats were randomly assigned to cardioprotective treadmill exercise or sedentary conditions before ischemia/reperfusion (ischemia 20 min, reperfusion 30 min) in vivo. Subsets of exercised animals received pharmacological inhibitors for mitoKATP (5-hydroxydecanoate) or sarc KATP (HMR1098) before ischemia/reperfusion. Analysis of digital ECG tracings revealed that mitoKATP inhibition blunted the antiarrhythmic effects of exercise, while sarc KATP inhibition did not (Fig. 19.2). Endogenous antioxidant enzyme activities for total, CuZn, and Mn superoxide dismutase, catalase, and glutathione peroxidase from ischemic and perfused ventricular tissue were not mitigated by IR, although oxidative stress was elevated in sedentary and mitoKATP-inhibited hearts from exercised animals. These findings suggest that the mitoKATP channel provides antidysrhythmic protection as part of exercise-mediated cardioprotection against ischemia/reperfusion, and the observed antidysrhythmic protection may be associated with preservation of redox balance in exercised hearts.
The Mitochondrial Calcium Uniporter
The role that intracellular calcium concentration plays in the induction of cardiac dysrhythmia has been extensively characterized [92, 93]. Early studies indicated that decreasing cytosolic calcium fluxes lowered the incidence of dysrhythmia [94], setting the stage for class IV antidysrhythmic agents that decrease dysrhythmias by lowering intracellular calcium.
However, the role of mitochondrial calcium fluxes in the generation of dysrhythmia is much less clear. Mitochondrial calcium homeostasis is believed to involve calcium influx into the matrix via the mitochondrial calcium uniporter (MCU), with the major efflux pathway being the mitochondrial sodium–calcium exchanger [95]. Attempts to decrease dysrhythmias by blocking MCU with ruthenium compounds have been mildly effective but only when given prior to ischemia. Pre-ischemic administration of both ruthenium red and Ru360 significantly decreased the incidence of ventricular fibrillation in anesthetized rats [96], and both ruthenium red and Ru360 effectively converted ventricular fibrillation to ventricular tachycardia, although neither compound led to the conversion to sinus rhythm [97].
Speculation regarding the mechanism whereby MCU can protect against dysrhythmia involves keeping matrix calcium concentrations low, ultimately leading to decreased probability of the opening of MPTP [96]. Although this mechanism may influence tissue survival, it seems unlikely to play a prominent role in dysrhythmogenesis, as blockers of the MPTP have not been particularly effective in preventing dysrhythmia. These findings are supported by experiments in myocytes, where the reversible collapse in Δψm induced during ROS-induced ROS release was indeed not prevented by either ruthenium red [41] or Ru360 [66].
At present, it is difficult to draw definite conclusions regarding the role of the MCU in dysrhythmogenesis due to the complicated effects of the ruthenium compounds on cellular calcium fluxes [98]. Ruthenium red has been shown to block calcium release from the SR [99] and L-type calcium channels [100], suggesting that the effects of this compound in preventing dysrhythmias may be mediated by a lowering of overall cellular calcium rather by directly acting on the mitochondrion [101]. Ru360 appears to be more specific for the MCU, but whole-heart experiments are confounded by permeability issues, with some investigators showing the Ru360 effectively enters cardiac cells 130 and others indicating that it is not permeable [102]. Consistent with their ability to reduce cytosolic calcium transients, both ruthenium compounds are potent negative inotropes at concentrations shown to protect against dysrhythmias [103], a potentially undesirable side effect when the overall clinical goal of administering the compound is to improve cardiac function. Future research with novel compounds that lack these pleiotropic/permeability issues will provide better insight into the role of the MCU in reperfusion dysrhythmias. One recent study suggested that pressure-puff-induced intracellular calcium releases were mediated by the mitochondrial efflux pathway, the mitochondrial sodium–calcium exchanger, which could potentially contribute to cardiac electrical dysfunction [104]. To date, studies examining mitochondrial calcium fluxes have mostly concentrated on the influx of calcium into the matrix via the MCU.
Mitochondrial Redox Status
The redox status of heart cells directly influences the cellular excitability. An oxidative shift in the cellular redox potential can promote action potential heterogeneity by modulating several different ion channels. Increased oxidation has been shown to directly activate sarcKATP channels [105], alter the inactivation kinetics of L-type calcium channels via increased calcium leak from the ryanodine receptor [106], and influence the state of channels on the mitochondrial inner membrane.
Bursts of ROS are observed within the first few minutes of reperfusion, when the propensity for dysrhythmia is extremely high [107]. In anesthetized rat, the prior administration of folic acid solution, amflutizole, superoxide dismutase, catalase, and superoxide dismutase plus catalase reduced the incidence of reperfusion-induced dysrhythmias and resultant mortality, caused by reperfusion after a transient period of coronary artery occlusion. Prior administration of soybean trypsin inhibitor significantly reduced mortality. In an isolated, perfused rat heart preparation with temporary coronary artery occlusion, addition of xanthine oxidase–hypoxanthine to the perfusion medium increased the incidence of reperfusion dysrhythmias and decreased the total duration of sinus rhythm during reperfusion. Further addition of superoxide dismutase or l-methionine increased significantly the total duration of sinus rhythm, suggesting that in the rat heart, xanthine oxidase may be involved in the genesis of reperfusion-induced dysrhythmias [108]. Several experiments have induced ventricular dysrhythmias under normoxic conditions with delivery of ROS bursts [109], and attempts to scavenge ROS with superoxide dismutase mimetics [110] or mitochondrial-targeted antioxidant peptides [111] were successful in decreasing the incidence of dysrhythmia. Future experiments that optimize effective delivery of ROS-scavenging agents to mitochondria clearly have significant potential in abrogating electrical dysfunction.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


