Mediastinal Paragangliomas and Pheochromocytomas
Raja R. Gopaldas
David C. Rice
Paraganglioma is a broad term generally used to describe a tumor that arises from chromaffin cells. Paragangliomas are rare tumors that arise from widely dispersed specialized neural crest cells associated with autonomic ganglia; they have the ability to secrete neuropeptides and catecholamines. Paraganglia are discrete collections of chromaffin cells distributed throughout the body. In other words, the etymology of the term paraganglioma stems from the description of extra-adrenal pheochromocytomas. Although the term mediastinal pheochromocytoma had been used quite frequently in the literature, the word pheochromocytoma should be specifically reserved to describe tumors of chromaffin cells arising primarily within the adrenal medulla. In this chapter we utilize the term paraganglioma to describe all pheochromatous tumors arising within the mediastinum, since they are by definition extra-adrenal. Owing to the extensive distribution of paraganglia throughout the body, paragangliomas have been reported in various locations: along the aorta and major vessels234 and in the heart, prostate, ovary, and urinary bladder.50 In addition to those originating from chromaffin cells, which are more common, paragangliomas also include tumors that arise from any type of chemoreceptor cells, the sympathetic chain, and the vagus.
Paraganglia are functionally classified as sympathetic or parasympathetic, based on cell of origin. Parasympathetic paraganglia do not secrete catecholamines and are chromaffin-negative.70 They are called chemodectomas and are more commonly associated with chemoreceptors located in the head and neck region,84,222 such as carotid body, glomus jugulare, glomus vagale, glomus ciliare, and glomus tympanum; occasionally, however, they may arise from mediastinal chemoreceptors such as the aortopulmonary glomus and anywhere along the intrathoracic course of the vagus.67
Functional mediastinal paragangliomas may be associated with physiologic effects similar to those of a pheochromocytoma. These effects may be life-threatening and clearly related to the hypersecretion of catecholamines.210 Patients undergoing surgery for a functional paraganglioma are subject to the effects of catecholamine hypersecretion preoperatively, massive catecholamine surges intraoperatively during tumor manipulation, and a catcholamine withdrawal phase with potential cardiovascular collapse right after the tumor is isolated. Several cardiovascular complications—such as left ventricular dysfunction,227 cardiogenic shock,29 pulmonary edema,110 and acute myocardial infarction150—have been reported. Telera230 described hemothorax as an initial presentation in patients with thoracic paraganglioma. The presentations of pheochromocytomas and functional paragangliomas are so dramatic and varied that they have been referred to as the “great mimics” in the medical literature.155
Functional paragangliomas account for 5% to 15% of all pheochromocytomas.252 The organ of Zuckerkandl is the source for more than 75% of functional paragangliomas, with the remaining 25% distributed in the thorax, mediastinum, and neck.247 Overall, <2% of paragangliomas occur in the chest.179 Although traditionally the incidence of extra-adrenal pheochromocytomas is believed to be 10%, McNichol,148 Disik,51 and Madani140 have quoted a higher percentage: 15% for adults and as high as 30% for children. Contrary to the traditional “rule of 10%” incidence of malignancy, Goldstein81 and Elder57 have reported a higher percentage of malignancy (between 26% and 40%) for functional extra-adrenal paragangliomas. Pommier187 demonstrated that overall and disease-free survival rates for malignant adrenal and extra-adrenal paragangliomas were similar and that the anatomic location had no prognostic value. The incidence of multicentricity of extra-adrenal paragangliomas ranges between 15% and 24%.78 Cardiac paragangliomas account for only 0.3% of all mediastinal neoplasms, half of which are functionally active. Some 10% of these tumors are malignant, as reported by Cane.30
Nonfunctional paragangliomas constitute a very small percentage of these tumors; the diagnosis is primarily correlated to symptoms secondary to local invasion and pressure effect. They do not cause hypercatecholaminemia. The effective diagnosis and management of the patient with a paraganglioma involves the close collaboration of endocrinologists, endocrine surgeons, anesthesiologists, geneticists, laboratory specialists, radiologists, oncologists, and pathologists. Paragangliomas are diagnosed in the following clinical settings: signs and symptoms related to catecholamine hypersecretion, mass effect symptoms, incidental finding on imaging, or family screening for hereditary paraganglioma.253
Embryology, Pathology, and Nomenclature
Chromaffin cells are derived from neuroectoderm and characterized by intense positive reaction of stored catecholamines with chromium salts, particularly potassium dichromate, giving a yellow-brown color on tissue sectioning.41 These cells are usually innervated by the preganglionic sympathetic nervous system and are capable of synthesizing and storing large quantities of catecholamines: adrenaline, noradrenaline, and epinephrine.42 The true chromaffin system, although closely associated with the sympathetic system, by itself comprises groups of cells topographically organized into the adrenal medulla, para-aortic bodies, paraganglia proper, carotid bodies, splanchnic nerves, prevertebral autonomic plexuses, and the small group of diffusely dispersed cells, paravertebral sympathetic ganglia. In essence chromaffin cells are distributed in almost all structures innervated by the autonomic sympathetic system, as described by Glenner and Grimley.77 The discovery of the enterochromaffin system and the amine-storing mast cells of the gut, pancreas, and liver, which also demonstrated chromaffin affinity, led to the belief that chromaffin affinity was not necessarily restricted to the sympathetic system. However, the common feature underlying all these cells is their amine-handling properties; hence the acronym APUD, for amine precursor uptake and decarboxylation, was introduced by Pearse182 to categorize these cells. Currently about 40 different types of APUD cells have been identified, which constitute the modern diffuse neuroendocrine system of the human body.156
Ever since the description of the formation of yellow-brown precipitate by reaction of catecholamine-producing tissues with chromic salts in 1902 by Kohn,117 the terms chromaffin reaction and chromaffin cells have been synonymous with the adrenal glands. However, works by Coupland43 recognized the fact that chromaffin staining was also characteristic of paraganglionic cells, which did not secrete catecholamines. In fact, the presence of catecholamine is essential for the chromaffin reaction, implying that although some cells did not secrete catecholamines, they still synthesized and stored them in cytoplasmic granules. This has been confirmed by electron microscopy. Histologically, functioning and nonfunctional paragangliomas are indistinguishable, as they both synthesize catecholamines and hence are chromaffin-positive. The only distinguishing factor is the level of circulating and excretory catecholamines and their metabolites, as recognized by McEwan et al.146 and Shapiro and Fig.210 Microscopically, the characteristic nesting pattern of cells termed “Zellballen” is unique to paragangliomas (Fig. 198-1). By immunohistochemistry, paragangliomas are positive for chromogranin, S-100 protein, and leu-encephalin, but they are negative for keratin.157 The presence of synaptophysin, chromogranin, and neuron-specific enolase in these cells indicates a neuroendocrine origin.104,216 Occasionally melanotic pigmentation has been identified in pheochromocytomas89 and cardiac paragangliomas,153 supporting divergent differentiation from neural crest cells expressing both pheochromatous and melanocytic features.35 The simultaneous expression of functional properties of two different types of neural crest cells in one tumor stresses the close relationship between all neural crest elements, as noted by Hofmann.95
The Paraganglia
Initially described by Zuckerkandl255 and Kohn,117 these cells are characterized by chromaffin staining, indicating the presence of catecholamine.41 The paraganglia are the predominant source of catecholamines in fetuses while the adrenal gland is developing. As the medullary component develops to a functional status, the paraganglial clusters involute, making them quite hard to detect unless they undergo neoplastic degeneration. As the fetus matures, there are still numerous persistent, minute paraganglia that are distributed throughout the body. Paraganglia contain two cell types. Type I cells possess numerous electron-dense granules that store large quantities of
catecholamines. Type II or satellite cells lack cytoplasmic granules and surround the type I cells partially or completely. Paraganglia are typically well vascularized. The type I cells are usually next to capillary fenestrations only separated by a basement membrane, allowing free flow of hormones into the circulation, as noted by Benedeczky.20 Although minute in size, they collectively have considerable metabolic importance and are a significant source of catecholamines in addition to the adrenal gland.
catecholamines. Type II or satellite cells lack cytoplasmic granules and surround the type I cells partially or completely. Paraganglia are typically well vascularized. The type I cells are usually next to capillary fenestrations only separated by a basement membrane, allowing free flow of hormones into the circulation, as noted by Benedeczky.20 Although minute in size, they collectively have considerable metabolic importance and are a significant source of catecholamines in addition to the adrenal gland.
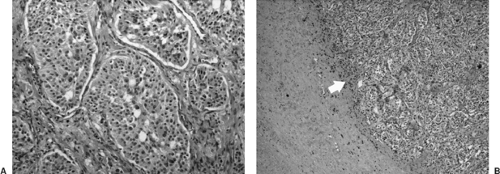 Figure 198-1. Photomicrograph of a paraganglioma that involved the left atrium. A: The tumor consists of typical large cellular trabeculae composed of mature pheochromocytes showing marked cellular and nuclear pleomorphism and some bizarre nuclear forms. B: The tumor can clearly be seen intimately involving the musculature of the left atrial wall without significant capsule formation (arrow). |
The World Health Organization (WHO) classification of endocrine tumors defines pheochromocytoma as a tumor arising from chromaffin cells in the adrenal medulla.224 Closely related tumors in extra-adrenal sympathetic and parasympathetic paraganglia are classified as extra-adrenal paragangliomas.122 A pheochromocytoma is an intra-adrenal sympathetic paraganglioma. While somewhat arbitrary, this nomenclature serves to emphasize important distinctive properties of intra-adrenal tumors that must be taken into account in clinical practice and research.234 Extra-adrenal paraganglia are divided into two categories: those related to the parasympathetic system and those connected with the sympathetic system. The former, usually nonchromaffin, lie in the head and neck, including the carotid body, intravagal, jugulotympanic, and mediastinal and aorticopulmonary paraganglia; the term chemodectoma is used exclusively for these tumors.120 The latter are chromaffin-positive tumors; they are associated with the peripheral sympathetic nervous system and secrete catecholamines in response to sympathetic neural stimulation. They lie in the para-axial region of the trunk close to the paravertebral and prevertebral ganglia, or in the connective tissue adjacent to pelvic organs, predominating along the thoracolumbar para-aortic region in the retroperitoneum.123,124 Because they are closely related to the sympathetic ganglia and their derivatives, these are properly described as paragangliomas by anatomic location.15 They are further subcategorized as functional or nonfunctional based on their ability to release the synthesized catecholamines into the systemic circulation, causing hormonal symptoms. These terms have been loosely utilized by a variety of authors; we prefer to categorize them based on the WHO classification as follows:
Pheochromocytoma—exclusively adrenal location.
Chemodectoma—arising from the parasympathetic ganglia. Nonchromaffin.
Functional paraganglioma—any extra-adrenal chromaffin-positive tumor that synthesizes and secretes catecholamines.
Nonfunctional paraganglioma—any extra-adrenal chromaffin- positive tumor that synthesizes but does not secrete catecholamines.
Although chemodectomas are common in the head and neck region, they have also been reported in the chest. Initially reported to be more common in the subpleural and parenchymal regions,213 these tumors are now believed to more often arise in the aortopulmonary region and the mediastinum, as reported by Nickels,168 Lacquet,125 Deodato,46 and Evora.62 There is probably a certain degree of underreporting owing to crossover of terms used for chemodectomas and nonfunctional paragangliomas. In addition, primitive childhood tumors of the sympatheticoadrenal system—such as neuroblastomas, ganglioneuroblastomas, and ganglioneuromas—may occasionally be erroneously reported as paragangliomas.
Extra-adrenal paraganglia have been classified into four anatomic groups by Glenner and Grimley77:
Branchiomeric (jugulotympanic, carotid body, laryngeal, subclavian, and aorticopulmonary); carotid body tumors are often referred to as chemodectomas.
Intravagal (at the level of the jugular or nodose ganglion); jugulotympanic tumors are sometimes referred to as glomus jugulare tumors.
Aortosympathetic.
Visceral autonomic.
The mediastinal paraganglia are predominantly concentrated in two locations: the aortosympathetic paraganglia are located along the sympathetic chain of the posterior mediastinum, and the aortopulmonary paraganglia reside along the great vessels and are part of the branchiomeric paraganglia (Fig. 198-2). Tumors arising from the aortosympathetic paraganglia will occur in the posterior mediastinum,193 whereas those arising from the aortopulmonary paraganglia will be intimately associated with the great vessels of the mediastinum.177 The visceral autonomic paraganglia, although rare, have recently gained attention. They arise within the cardiac chambers, particularly the left atrium, and pose diagnostic and therapeutic challenges. These are discussed below, under “Cardiac Paragangliomas.”
Aortopulmonary paraganglia function as chemoreceptors, like those found in the carotid body. They sense fluctuations in blood pH, oxygen, carbon dioxide partial pressure, and temperature, and influence homeostasis by altering ventilation and heart rate.121 The aortopulmonary paraganglia are characteristically found in five different locations: (a) between the ascending aorta and pulmonary trunk (either anteriorly or posteriorly), adjacent to the aortic root (also known as the coronary paraganglia); (b) within the groove between the ductus arteriosus and the pulmonary artery (also known as the pulmonary paraganglia); (c) between the origin of the right subclavian and right common carotid arteries; (d) between the origin of the left subclavian and left common carotid arteries; and (e) caudad to the left subclavian artery adjacent to the aortic arch. The latter four are collectively referred to as subclavian–supra-aortic paraganglia.123 Bird and Seiler24 have noted that all of these paraganglia are capable of storing intracellular catecholamines. Functional paraganglia that store and secrete catecholamines may occur in any of the four paraganglia groups. In addition, islands of carotid body–type chemoreceptive tissues are also found in the pericardium, as Shapiro et al.211 and Dresler and coworkers53 have reported.
Clinical Presentation
The clinical presentation of paragangliomas can be varied and ranges from local pressure symptoms, especially in the case of nonfunctional tumors, to symptoms related to the systemic effects of secreted catecholamines and their metabolites in functional tumors. In many instances, complications arising from hypercatecholaminemia are the primary reason medical attention is sought for these patients, which then uncovers an underlying functional paraganglioma.
Frequently the clinical presentations of these tumors are ill defined. Hypertension is the most common symptom in 82% of
cases with functioning paraganglioma, as noted by Goldstein.82 Indeed, paragangliomas may account for up to 0.5% of all cases of hypertension, and elevated blood pressure may be the only initial presentation of these tumors, as reported by Chao.33 In one series of 236 patients with benign paragangliomas (of all locations), catecholamine hypersecretion was found in 40 of 128 screened patients (31%) and 38 were hypertensive.60
cases with functioning paraganglioma, as noted by Goldstein.82 Indeed, paragangliomas may account for up to 0.5% of all cases of hypertension, and elevated blood pressure may be the only initial presentation of these tumors, as reported by Chao.33 In one series of 236 patients with benign paragangliomas (of all locations), catecholamine hypersecretion was found in 40 of 128 screened patients (31%) and 38 were hypertensive.60
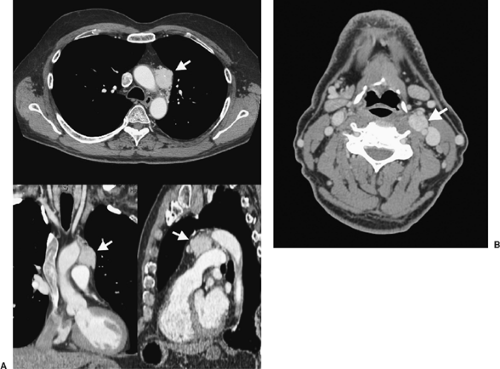 Figure 198-2. CT scan of a patient with branchiomeric (aorticopulmonary) paraganglioma (arrow). Histologic diagnosis was confirmed with percutaneous CT-guided needle biopsy. B: The patient also had a left-sided carotid body tumor, which appeared as a contrast-enhancing mass situated just above the bifurcation of the left carotid artery (arrow). He was managed nonoperatively and had stable disease for >5 years. |
In addition to hypertension, secretion of vasoactive catecholamines may lead to other symptoms of catecholamine excess, such as headache, palpitations, sweating, nausea, shortness of breath, weakness, myocardial infarction, flushing, weight loss, visual changes, stroke, coma, arrhythmias, and psychosis. Ventricular tachycardia can be a life-threatening initial presentation in functional paragangliomas, as reported by Michaels,152 Petit,184 and Li.133 Some types of cardiac paraganglia are believed to derive their blood supply from the coronary vasculature and may manifest as atypical angina or even myocardial ischemia.
Both sexes are equally affected. The peak incidence is in the third and fourth decades of life, as reported by Freier,69 and children are occasionally affected, according to DiStefano49 and Petit et al.184
Workup of young patients with mediastinal paraganglioma should promptly include evaluation of the upper GI tract and chest wall to exclude the presence of Carney’s triad. First described in 1979 by Carney,31 the triad comprises gastric epithelioid leiomyosarcoma, functioning extra-adrenal paraganglioma, and pulmonary chondroma, with patients usually presenting in the second or third decade of life. As two of the three components of the triad are potentially lethal, it is essential that any young patient with a functioning paraganglioma be thoroughly evaluated for the presence of other tumors.31,44
Confirmation of the diagnosis depends on the functionality of the tumor. Tissue diagnosis is best avoided in functional tumors, as this can precipitate a hypertensive crisis. In addition, most of these tumors are highly vascular and are liable to cause bleeding complications. In the presence of elevated catecholamines and anatomic localization of the tumor, functional studies may be utilized and a tissue diagnosis is generally not necessary. If tissue biopsy has been obtained, several markers help in establishing the diagnosis. Paragangliomas simultaneously coexpress catecholamines and different peptides. Pavai181 has suggested that the diagnosis is essentially confirmed by S-100 and chromogranin positivity. Chromogranin by itself has a low sensitivity, but neuron-specific enolase (NSE) and chromogranin together have a sensitivity that approaches 100%, as reported by Kliewer.115 It has been observed that undifferentiated tumors with malignant features are less likely to express characteristic tumor markers, whereas more differentiated tumors are more prone to express them. In addition paragangliomas express a variety of neuropeptides, the more common ones being metencephaline (73%–75%) and leu-encephaline (50%–76%),
as reported by Warren.244 Parasympathetic paragangliomas are usually chromaffin-negative.2
as reported by Warren.244 Parasympathetic paragangliomas are usually chromaffin-negative.2
Hereditary Syndromes Associated with Paraganglia
Several syndromes have been associated with paragangliomas, including neurofibromatosis type 1 (NF1), Von Hippel–Lindau disease (VHL), and hereditary paraganglioma syndromes 1 through 4 (PGL1–4). It is important to understand these syndromes, as a chromaffin-positive tumor may be the only manifestation of the underlying genetic mutation. Almost all of these associated syndromes are autosomal dominant.
Neurofibromatosis type 1 is an autosomal dominant disorder caused by inactivating mutations of neurofibromin, a tumor suppressor gene. Clinically it manifests itself with café au lait spots, axillary freckling, multiple neuromas, iris hamartomas (known as Lisch nodules) and chromaffin tumors. The majority of chromaffin tumors in these patients appear in the fourth decade of life. In many circumstances the diagnosis of NF1 is made during childhood, as the typical skin lesions precede the appearance of paragangliomas/pheochromocytomas. Hence the chance that NF1 could manifest as an apparent sporadic paraganglioma is quite rare, as reported by Gutmann.87 Overall, extra-adrenal sympathetic paragangliomas are present in 6% of patients with NF1, the majority being benign, as noted by Walther.242
VHL disease is an autosomal dominant syndrome, clinically associated with both benign and malignant tumors: renal and testicular tumors and pancreatic cysts, islet cell tumors, retinal angiomatosis, cerebellar hemangioblastoma, chromaffin tumors, and renal cell carcinoma.94 Some 50% of patients with VHL present with a catecholamine-producing tumor. About 2% of these are thoracic sympathetic paragangliomas. More than 90% of functional chromaffin tumors in patients with VHL are benign, with a higher incidence of malignant behavior quoted among paragangliomas of extra-adrenal origin.242 These catecholamine-producing tumors in patients with VHL mutation may manifest as the first or only evidence of the disease.
Hereditary paragangliomas account for about 30% of cases of paragangliomas, highlighting the importance of family history in young patients who present with paragangliomas, as reported by Van Baars.237 Hereditary paragangliomas are characterized by an earlier age of onset and more severe form of disease. Four different loci have been implicated giving rise to hereditary paraganglioma syndromes PGL1 through PGL4: PGL1 on 11q23, PGL2 on 11q13, PGL3 on 1q21, and PGL4 on 1p36, as studied by Favier.69 Hereditary paragangliomas are inherited in an autosomal dominant pattern with incomplete penetrance and variable expressivity. Until the year 2000, only two syndromes (VHL and NF1) were known to be associated with extra-adrenal paragangliomas. The recent identification of germline mutations in the SDH genes encoding succinate dehydrogenase in patients with familial paraganglioma has highlighted a primary role of mitochondrial deficiency in carcinogenesis, as reported by Eng.59 The discovery of the first deleterious mutations in the SDHD gene for PGL1 by Baysal et al.18 and subsequent mutations in SDHC (PGL3) by Niemann169 and SDHB (PGL4) by Astuti8 have resulted in significant changes in the counseling and genetic workup of patients with hereditary paragangliomas. Several mutations in these genes have subsequently been reported in patients with hereditary PGL and sporadic cases as well by Baysal.16 SDH gene mutations are also frequently encountered in patients with an apparently sporadic form of paragangliomas.
Most of the hereditary paraganglioma syndromes are associated with head and neck parasympathetic paragangliomas (chemodectomas) that usually remain nonfunctional. Only PGL1 and PGL4 have been found to be associated with functional types of sympathetic paragangliomas. PGL 3 mutations of succinate dehydrogenase Subunit C are associated with benign nonfunctional head and neck parasympathetic paragangliomas. So far there has been no association with functional sympathetic paragangliomas or pheochromocytomas as observed by Schiavi.217 The causative gene for PGL2 remains to be identified. Currently this has been exclusively associated with parasympathetic nonfunctional paragangliomas of head and neck as reported by Baysal.17 The association with mediastinal tumors is rare.
MEN1 and MEN2 are commonly associated with pheochromocytomas but rarely with thoracic paragangliomas. Although MEN2 is associated with pheochromocytomas, the incidence of paragangliomas is very low, most of which have been in the periadrenal area or after the primary adrenal tumor has been resected. This has been attributed to ectopic rests of adrenal tissue or secondary to tumor recurrence or spillage as noted by Nilsson.170 Hence, thoracic paragangliomas are almost never associated with MEN2 syndrome. Neumann et al.164 and Gimenez-Roqueplo et al.74 reported that about 50% of patients with extra-adrenal tumors were found to harbor germline mutations, most commonly of succinate dehydrogenase subunits B and D, thereby necessitating genetic testing as a part of the initial workup of patients presenting with functional paragangliomas. The presence of multicentric tumors and head and neck parasympathetic paragangliomas should also raise the possibility of underlying genetic syndrome, prompting early genetic counseling and testing.
Reports by Bauters14 and Neumann164 have suggested that the likelihood of finding a hereditary basis for functional paragangliomas is increased when age of onset is young (≤18 years). About half of those presenting <20 years have hereditary disease. The first line of genetic testing for those with mediastinal paragangliomas should be for Von Hippel–Lindau germline, as the incidence is about 74% in patients <20 years of age. The next in line are SDHB and SDHD mutations. The incidence of germline mutation progressively decreases as the age of presentation increases. For patients >50 years who present with an apparently sporadic paraganglioma, the probability of having a VHL, SDHB, or SDHD mutation is <2%, making genetic testing unnecessary and difficult to justify.
The Catecholamine Syndrome
Hypertension is the primary underlying feature of catechola- mine-secreting paragangliomas. The presence of severe and symptomatic paroxysmal hypertension should always generate suspicion of a catecholamine-secreting pheochromocytoma, as noted by Goldstein.81 Not all patients with a functioning paraganglioma will necessarily present with paroxysmal episodes, as many patients have persistent hypertension without episodic increases in blood pressure. In addition, there have been reports
of patients with functioning paragangliomas without clinical evidence of hypertension. Catecholamine-producing tumors may arise in the adrenal medulla (pheochromocytomas) or in extra-adrenal chromaffin cells (secreting paragangliomas). The estimated overall prevalence (based on autopsy studies) is 0.05% to 0.12%,5 and an estimated prevalence in unselected patients with hypertension is 0.2% to 0.6%.155 Hence it is important to suspect, confirm, localize, and resect these tumors, because the hypertension associated with pheochromocytoma is curable, and these patients are at risk for a lethal paroxysm.
of patients with functioning paragangliomas without clinical evidence of hypertension. Catecholamine-producing tumors may arise in the adrenal medulla (pheochromocytomas) or in extra-adrenal chromaffin cells (secreting paragangliomas). The estimated overall prevalence (based on autopsy studies) is 0.05% to 0.12%,5 and an estimated prevalence in unselected patients with hypertension is 0.2% to 0.6%.155 Hence it is important to suspect, confirm, localize, and resect these tumors, because the hypertension associated with pheochromocytoma is curable, and these patients are at risk for a lethal paroxysm.
Functional paragangliomas may secrete either norepi- nephrine or epinephrine but predominantly release norepi- nephrine. Rarely, they may secrete dopa and/or dopamine. Approximately half of patients have permanent hypertension, the rest suffer from paroxysmal hypertension, and there is no clear reason as to why patients may present in one form or the other, as noted by Reisch.196 Paroxysms may occur on a background of sustained hypertension, whereas others can have normal blood pressure between paroxysms, as reported by Lenders.130 The frequency of paroxysms is quite varied: 75% of affected patients suffer from attacks weekly, others several times per day or just once every few months. Paroxysms occur suddenly and unexpectedly. In 80%, they last <1 hour, then subside gradually and lead to exhaustion.141 The attacks are characterized by headache (50%), sweating (50%), and palpitations (50%–60%). However, recent studies showed that this classic triad occurs more rarely (15%–24%) than usually assumed, as observed by Baguet.11 Even hypertension appears to be less frequent (60%–70%) than stated in earlier studies by Mannelli.142 Normotensive courses are more frequent in familial pheochromocytomas, as observed by Plouin.186
Because catecholamines can inhibit peristalsis, functional paragangliomas may be associated with severe constipation, pseudo-obstruction, or ileus, as reported by Murakami.160
Mesenteric artery vasoconstriction due to hypercatecholaminemia may result in ischemic enterocolitis with intestinal necrosis. Because thoracic paragangliomas are rare tumors, most of the complications reported in the literature pertain to excessive catecholamine secretion from adrenal pheochromocytomas. These systemic effects, however, hold true for functional paragangliomas anywhere in the body. Bowel perforation,107 stroke, dilated cardiomyopathy,134 sick sinus syndrome,85 and a variety of other complications from catecholamine secretion have been reported. The intense vasoconstriction also leads to volume contraction and hemoconcentration, which has an important bearing on the preoperative management of patients with catecholamine-secreting tumors. In any case of sustained paroxysmal hypertension or paradoxical hypertension despite antihypertensive therapy, especially during therapy with beta-blockers, the diagnosis of pheochromocytoma has to be kept in mind and ruled out. New onset of hypertension under tricyclic antidepressive medication, severe symptomatic hypotension when starting therapy with alpha blockers, or severe retinopathy in newly diagnosed hypertension may suggest functional catecholamine-producing tumor.
Neuroendocrine Effects
In addition to a catecholamine excess, pheochromocytomas have been found to secrete neuropeptide Y or chromogranin A. Rarely, they may release vasointestinal peptide, serotonin, calcitonin, parathyroid hormone–related protein, adrenocorticotropic hormone (ACTH), neuron-specific enolase, or interleukin-6, as noted by Yeo.252 Production of vasoactive intestinal polypeptide by functional chromaffin tumors is extremely rare. Since the first report in 1975 by Loehy et al.,137 only 16 cases of adrenal pheochromocytoma associated with the watery diarrhea, hypokalemia, and achlorhydria (WDHA) syndrome have been reported, as summarized by Ikuta.97 Some of these presentations have not been reported yet for thoracic functional paragangliomas because these are unusual manifestations in an extremely rare tumor to begin with. Excessive insulin production is extremely rare, as reported by Fujino71 and Uysal.236 Although erythropoietin has not been directly implicated, refractory polycythemia has been reported by Imai.99 Terada231 and associates demonstrated a lack of elevation in erythropoietin levels in patients with functioning paragangliomas, favoring the volume contraction theory as the more likely cause of polycythemia. Systemic amyloidosis was reported by Rey197 and hypercalcemia due to parathormone related peptide production was reported by Loh.138 A wide range of peptide hormones may be present within the tumor and identified by immunohistochemistry without necessarily giving rise to clinical syndromes, as reported by Shapiro and Fig.210
Mass Effects
As Smit and associates221 reported, functional paragangliomas may, in addition to the effects of catecholamine hypersecretion, exert local pressure effects, whereas nonsecreting paragangliomas present only with local mass effects. The latter may thus be larger at the time of presentation. Many are discovered incidentally on chest radiography performed for other reasons. Within the chest, lesions in the paravertebral sulci may cause nerve root compression, spinal cord compression, or both if they invade the neural canal; Horner’s syndrome may be observed if the tumor interrupts the thoracocervical sympathetic outflow. Lesions arising close to the coronary arteries may compress them and cause angina. Large lesions may compress lung parenchyma. Mediastinal lesions in the visceral compartment may compress structures, particularly low-pressure thin-walled veins or cardiac atria, or may affect the recurrent laryngeal nerve. Left atrial tumors are associated with compressive symptoms obstructing pulmonary venous inflow into the left atrium.
Metastatic Disease
The overall frequency of malignancy in adrenal paragangliomas had been reported to be 10%. Extra-adrenal primary tumors appear to have a greater metastatic potential than adrenal medullary tumors, as pointed out by Shapiro211 and Bravo.26 The most common metastatic sites for chromaffin-cell tumors are local lymph nodes, bone (50%), liver (50%), and lung (30%), as reported by Bravo26 and Loh et al.139 Malignant lesions have a propensity to spread to bone, as noted by Shapiro et al.,210 Scharf,205 and Lamy127 and their colleagues, where the deposits may cause bone pain and pathologic fractures, but they are often asymptomatic, as reported by Heindel and colleagues.91 Rarely,
metastases to brain, skin, or serous cavities have been described. Metastases may become clinically evident after long latent intervals following apparent cure; thus lifelong follow-up is recommended. Persistent postoperative symptoms in patients with chromaffin-cell tumors in the absence of radiologically evident residual tumor may suggest the presence of occult metastases.103
metastases to brain, skin, or serous cavities have been described. Metastases may become clinically evident after long latent intervals following apparent cure; thus lifelong follow-up is recommended. Persistent postoperative symptoms in patients with chromaffin-cell tumors in the absence of radiologically evident residual tumor may suggest the presence of occult metastases.103
The histologic findings do not permit definite diagnosis of malignancy in chromaffin-cell tumors; this still remains a controversial topic. Several possible correlates have been suggested, such as tumor weight >80 g and high tumor concentration of dopamine by John et al.,103 tumor size >5 cm (75% prevalence of malignancy) by Goldstein et al.,81 or the presence of confluent tumor necrosis (a common feature in larger tumors). None of these, however, can conclusively predict malignant behavior.
When there is recurrence of disease, biochemical evidence of excess catecholamine production usually precedes the clinical manifestations of catecholamine excess.246 Detection of malignant characteristics primarily relies on follow-up, as there are no reliable biochemical, clinical, or histologic features that reliably predict malignancy, as noted by Pommier.187 Invasion of surrounding tissues, spread to lymphatics, and presence of metastatic disease are the only definitive predictors of malignancy, as described by Sandur,204 Whalen,247 and Goldfarb.78 Although none of these are diagnostic, the presence of nuclear atypia, vascular invasion, or tumor necrosis may suggest a malignant potential, as highlighted by Shapiro.211 The absence of these histologic findings does not exclude malignancy, as seemingly benign lesions may metastasize widely, with the only proof of malignancy dependent on the unequivocal presence of metastatic disease or local infiltration of surrounding structures.
As the distinction between malignant and benign tumors still remains difficult, there is a pressing necessity to identify markers that can reliably predict tumors with malignant potential. As pointed out by Herrera et al.,92 DNA ploidy correlates poorly with malignant potential. Kumaki et al.119 have suggested MIB-1 immunostaining as a useful adjunctive marker to predict malignant behavior in pheochromocytomas and paragangliomas. Recent molecular studies have shown that the prevalence of malignancy among paragangliomas is higher; especially those associated with succinate dehydrogenase subunit B gene mutations, as pointed by Chrisoulidou.36 Genetic profiling of tumor cells has recently been proposed by Thouennon et al.232 as a method of distinguishing malignant adrenal pheochromocytomas. Lower expression of key genes encoding peptide processing and activation factors—such as neuroserpin, glutaminyl-peptide cyclotransferase, peptidylglycine alpha-amidating mono-oxygenase—has been associated with greater malignant potential. The gamma-tubulin gene, which encodes a constituent of centrosomes, has been noted to be significantly overexpressed in malignant pheochromocytomas, while the genetic expression of occludin, a major component of junctions, is repressed in malignant tumors, suggesting a possible diminution of cell-to-cell contacts and an increased permeability in malignant adrenal pheochromocytomas. These findings are yet to be further explored in the realm of extra-adrenal paragangliomas. For all practical purposes paragangliomas are to be regarded as malignant and surgical principles are heavily reliant on this fact. The only absolute criterion for malignancy, however, is the presence of metastases to sites where chromaffin tissue is not usually found. The lack of firm predictors of malignancy coupled with the variable course of this rare disease makes lifelong follow-up of patients with chromaffin-cell tumors mandatory.
Biochemical Diagnosis
All patients with suspected functional paragangliomas should undergo biochemical testing. This includes patients with suggestive paroxysmal signs or symptoms; those with recent, therapy-resistant, or volatile hypertension; and those with a hereditary predisposition for a paraganglioma. Because of the low prevalence of these tumors, biochemical screening for the tumor in asymptomatic patients with hypertension is not indicated.
Two important factors guiding biochemical diagnosis are (a) to investigate signs and symptoms that suggest the presence of a functional paraganglioma and (b) to determine catecholamine hypersecretion in patients presenting with a mediastinal mass suspicious of a paraganglioma.
Nonfunctioning paragangliomas are unlikely to be diagnosed with biochemical testing unless a tissue sample is obtained that reveals the presence of chromaffin-positive cytoplasmic granules. The best screening tests for initial assessment of functioning chromaffin-cell tumors are the measurement of plasma free and urinary fractionated metanephrines, as reported by Lenders.129 Extra-adrenal chromaffin tumors are more likely to produce norepinephrine, as noted by van der Harst.238 Plasma analysis for diagnosis includes measurement of catecholamines, norepinephrine and epinephrine, and free metanephrines, normetanephrine, and metanephrine. Urinary measurements include metanephrine, normetanephrine, and vanillyl mandelic acid (VMA). Based on work by Lenders,129 sensitivities are highest for measurements of plasma free metanephrines (99%), followed closely by urinary fractionated metanephrines (97%). When used in combination, sensitivities of both the above tests considerably exceeded those for urinary catecholamines at 86%, plasma catecholamines at 84%, and urinary total metanephrines at 77%. The highest specificities were 95% for tests of urinary VMA and 93% for tests of urinary total metanephrines. Specificities of all tests were higher in patients tested for hereditary chromaffin tumors than for sporadic tumors. To minimize the risk of missing a patient with paraganglioma, clinicians often use multiple biochemical tests during the initial diagnostic workup of patients with suspected tumors. Plasma free metanephrines constitute the best test for excluding or confirming functional paragangliomas and should be the test of first choice for diagnosis of the tumor. A negative test result virtually excludes functioning chromaffin tumors. In many cases the diagnosis is usually accomplished fairly easily as the levels are remarkably elevated and the task of localization becomes more of a challenge. The molecular basis of metanephrine measurement is that chromaffin-cell tumors contain catechol-O-methyl-transferase, which metabolizes adrenaline and noradrenaline to metanephrine and normetanephrine respectively. However, one must be cognizant of the fact measurement of metanephrines may fail to identify tumors that secrete small amounts of catecholamines and those that exclusively produce dopamine, as pointed by Eisenhofer et al.55
In dopamine-secreting tumors, measurement of plasma dopamine or its O-methylated metabolite, methoxytyramine, provides higher diagnostic accuracy than urinary dopamine. To minimize false positive results, medications known to interfere
with catecholamine metabolism (tricyclic antidepressants, phenoxybenzamine, and labetalol) should be avoided if possible, as noted by Eisenhofer et al.56 Obtaining blood samples from seated rather than supine patients may be more practical for phlebotomists, but the sympathoadrenal activating effects of upright posture may compromise the diagnostic accuracy of measurements of plasma metanephrines. Use of reference intervals established from samples obtained from seated patients results in threefold increase in false-negative test results and loss of diagnostic sensitivity. Blood samples for diagnosis of chromaffin-secreting tumors should be obtained only with patients in the supine position.
with catecholamine metabolism (tricyclic antidepressants, phenoxybenzamine, and labetalol) should be avoided if possible, as noted by Eisenhofer et al.56 Obtaining blood samples from seated rather than supine patients may be more practical for phlebotomists, but the sympathoadrenal activating effects of upright posture may compromise the diagnostic accuracy of measurements of plasma metanephrines. Use of reference intervals established from samples obtained from seated patients results in threefold increase in false-negative test results and loss of diagnostic sensitivity. Blood samples for diagnosis of chromaffin-secreting tumors should be obtained only with patients in the supine position.
Biochemical studies are inadequate to either predict malignancy or distinguish benign from malignant lesions, as noted by Ahlman.3 Plasma chromogranin A (CgA) has also been used for the diagnosis and prediction of malignant behavior in chromaffin-cell tumors by O’Connor,172 and neuron-specific enolase has been advocated as a screening marker for malignant behavior by Oishi and Sato.175 A direct relation of ACTH overexpression and malignant behavior was reported by Moreno et al.159 Other measurements, such as platelet norepinephrine measurements, have a sensitivity of 93%, as reported by Guller et al.86 Platelet measures have the presumed advantage of maintaining a fairly consistent level over a period of time despite rapid fluctuation in plasma concentration of catecholamine derivatives.
Suppression and Stimulation Tests
Plasma norepinephrine concentrations typically are high and fail to decrease normally by 3 hours after oral administration of 300 μg of clonidine, which decreases the release of norepi- nephrine from sympathetic nerve endings but not from tumors. The failure of clonidine to suppress norepinephrine levels therefore is a positive, or abnormal, result. The clonidine suppression test, first described by Bravo in 1981,25 is accepted as a diagnostic test in patients with elevated baseline norepinephrine levels. However, the clonidine suppression test is not specific enough when it is used as a single test. False-positive and false-negative results have been obtained from clonidine suppression testing, since some patients without catecholamine-secreting tumors can fail to suppress plasma norepinephrine levels after clonidine administration, and some with paragangliomas can have normal levels of norepinephrine after a clonidine suppression test, as pointed out by Grossman.85
Several stimulation tests have been proposed in the diagnostic evaluation of functional chromaffin tumors, including pressor responses to sympathomimetic agents and glucagons. These can produce dangerous increases in pressure in patients with functional tumors; hence stimulation tests involving measurements of pressor responses are no longer recommended.
Imaging
Preoperative localization is essential in the management of paragangliomas of the thorax. Surgical planning and determination of operative approach is heavily dependent on precise anatomic localization and relationship of the tumor to the surrounding structures.213 Two types of imaging are utilized for functional paragangliomas: (a) anatomic imaging to locate the extent of the tumor and relation to surrounding structures and (b) functional imaging, which is utilized as screening for metastatic disease, staging, workup, and for localizing the tumor in patients with biochemically confirmed diagnosis.
Anatomic imaging can include a variety of tests, such as computed tomography (CT), magnetic resonance imaging (MRI), transthoracic echocardiography, transesophageal echocardiography, and chest radiography with barium swallow.
Functional studies include metaiodobenzylguanidine (MIBG) scans and somatostatin receptor scintigraphy. The choice of test depends on the clinical presentation, location and functional status of the tumor.
Chest Radiography
Chest radiography is a basic imaging modality for any intrathoracic lesion; however, it is helpful only for localizing tumors in the parenchyma or paravertebral sulci. Diagnosis of smaller mediastinal tumors is usually impossible with chest radiographs. Barium swallow is helpful in tumors arising in the retrocardiac regions causing impingement of the esophagus. This especially holds true for tumors arising from the left atrium, which is anatomically in close relation to the esophagus. Visceral type of paragangliomas have a preponderance for occurrence in the left atrium, which may lend to their recognition by barium contrast esophagogram, as reported by Tekin et al.229 (Fig. 198-3).
Thallium-201 Scans and Myocardial Function Studies
Tumors arising within the pericardium may parasitize the coronary vasculature and cause an atypical type of anginal pain. Patients who present with such lesions may benefit from thallium scintigraphy, as suggested by Orringer et al.,176 to evaluate regions of myocardium that may be potentially compromised. In addition, patients with complications of hypercatecholaminemia, such as cardiomyopathy, would benefit from gated blood pool studies to determine myocardial function and ejection fraction before embarking on a surgical procedure. Based on thallium scans, resolution of myocardial dysfunction after resection of the functional chromaffin tumor has been noted by Gatzoulis.73 Thallium scans are also useful as an adjunct to MIBG scans when tumors are in close proximity to the myocardium, as they can help in the recognition of intracardiac functional tumors, as pointed by Basoglu.13 In cases where the MIBG scan is equivocal, direct thallium or thallium-technetium subtraction scans have demonstrated the functional chromaffin tumors, as noted by Nakajo162 and Wenisch,245 respectively.
Echocardiography
Echocardiography has a definite role in the investigation of visceroautonomic types of paragangliomas, which are frequently intrapericardial lesions. The visceroautonomic paraganglia are a poorly characterized group composed of microscopic cell groups found in the interatrial septum of the heart. The visceroautonomic type of intracardiac paraganglioma is typically known to occur within the left atrium on the septal aspect.
Transesophageal echocardiography (TEE) has been used in the workup of cardiac paragangliomas, as reported by Tekin229 and Maxey.145 On echocardiography, paragangliomas usually appear as large, echogenic left atrial masses; they have been mistaken for myxomas, as pointed by Turley.235 Unlike myxomas, however, paragangliomas have a broad base of attachment. Although the use of TEE provides valuable information, one must exercise caution, especially with functional tumors, as a hypertensive crisis can be precipitated by noxious stimuli from the probe. In addition, intraoperative TEE monitoring is essential for patients undergoing a major cardiac procedure for resection of these tumors.
Transesophageal echocardiography (TEE) has been used in the workup of cardiac paragangliomas, as reported by Tekin229 and Maxey.145 On echocardiography, paragangliomas usually appear as large, echogenic left atrial masses; they have been mistaken for myxomas, as pointed by Turley.235 Unlike myxomas, however, paragangliomas have a broad base of attachment. Although the use of TEE provides valuable information, one must exercise caution, especially with functional tumors, as a hypertensive crisis can be precipitated by noxious stimuli from the probe. In addition, intraoperative TEE monitoring is essential for patients undergoing a major cardiac procedure for resection of these tumors.
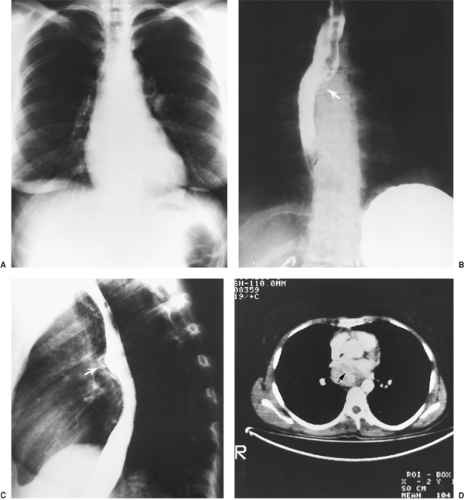 Figure 198-3. The need for pericardiotomy for midmediastinal pheochromocytomas. A: Normal chest radiograph in a 19-year-old woman. Venous sampling of plasma catecholamine levels localized her pheochromocytoma to the midmediastinum. She underwent an exploratory thoracotomy at another hospital, and no tumor was found. Posteroanterior (B) and lateral (C) views via esophagography show an extrinsic displacement of the esophagus in the subcarinal region by the tumor. Surgical clips from the initial exploration are visible in the subcarinal region (arrows). D: Chest CT scan shows a left atrial pheochromocytoma (arrow), which was localized with a 131I–metaiodobenzylguanidine scan. The tumor was soft and flattened in its intrapericardial location; failure to perform a pericardiotomy at the thoracic exploration led to the surgeon’s inability to locate it.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|