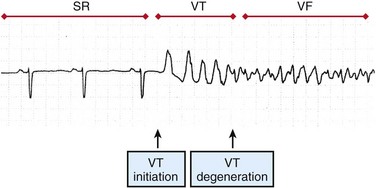
48 Sudden cardiac death (SCD) causes an estimated 300,000 deaths in the United States alone.1 Ventricular tachycardia (VT) often precedes the onset of ventricular fibrillation (VF). VF causes approximately one-third of sudden cardiac deaths.2 Patients at high risk for SCD may be implanted with an implanted cardioverter-defibrillator (ICD), but the largest group of victims of SCD does not have risk factors that place them in the high-risk category as candidates for ICD implantation.3 To develop more effective treatments for VT and VF, the mechanisms of VT and VF onset and maintenance must be understood. Although cases of idiopathic VF have been reported, most patients with VT and VF have a substrate that increases the probability of reentry. Two primary conditions lead to the onset of VT: ectopic foci and stable reentrant circuits. Ectopic foci due to triggered activity or abnormal automaticity may lead to VT. VT may be hemodyamically stable or unstable and often self-terminates, whereas VF is almost always fatal if not treated by administration of defibrillation shocks within minutes of VF onset. Ventricular tachyarrhythmias often progress from premature ventricular complexes (PVCs) to runs of VT, and finally to VF (Figure 48-1). Ventricular tachycardia and heart failure lead to action potential duration (APD) shortening, and rapid activation rates may lead to intracellular calcium overload. High intracellular calcium and APD shortening promote triggered activity and the initiation of VF.4 Figure 48-1 Surface ECG recording of the transition from normal sinus rhythm (SR) to ventricular tachycardia (VT) and eventually to ventricular fibrillation (VF). (Reprinted with permission from Weiss JN, Garfinkel A, Karagueuzian HS, et al: Chaos and the transition to ventricular fibrillation: A new approach to antiarrhythmic drug evaluation. Circulation 99:2819-2826, 1999.) Focal sources that may lead to VT include triggered activity and abnormal automaticity. Focal sources may be the source of repetitive rapid ectopic firing or they may lead to disruption of the normal conduction pathways and the breakup of cohesive wave fronts and establishment of reentrant circuits (Figure 48-2, A). Delayed afterdepolarizations (DADs) cause a rise in resting potential during diastole. If the transmembrane potential rises above the activation threshold, a new action potential may be launched. DADs are traditionally linked to intracellular calcium overload and thus are exacerbated by the rapid heart rates seen in VT and VF. Early afterdepolarizations (EADs) may occur during the plateau (phase 2) or repolarization (phase 3) phase of an action potential. EADs are often associated with bradycardia or slow heart rate and have been linked to numerous ion channel disturbances including L-type calcium, rectifier potassium, and late sodium currents. The sodium/calcium exchanger current is thought to play a critical role in the development of EADs. Chua et al performed optical mapping of tachycardia-induced heart failure and demonstrated that heterogeneous up-regulation of apamin-sensitive K+ current increases sensitivity to intracellular calcium.5 This leads to heterogeneous APD shortening and possibly to late phase 3 EADs in the context of high heart rate or post-defibrillation recovery. Stretch activation may play a role in ventricular arrhythmias, particularly in the context of severe heart failure and volume overload.6 Abnormal automaticity consists of abnormal spontaneous firing action potentials that are not coupled to previous activations. Figure 48-2 The mechanisms of VT and VF are demonstrated in 2D simulations of transmembrane voltage Stable reentrant circuits around an anatomic anchor such as a scar or a large vessel may lead to sustained VT (see Figure 48-2, A). A section of unexcitable post-infarct scar or a large vessel such as the aorta or the right ventricular (RV) outflow tract may form a pathway for a stable reentrant circuit. Even without an unexcitable core, simulations have shown that a reentrant circuit may be formed around a functional core rather than an anatomic core (see Figure 48-2, A). The center of the circuit may remain excitable but unexcited as the spiral wave circles around the core. Large reentrant circuits that encircle the entire ventricles have been shown to form as well, especially with cardiac dilatation or conduction slowing. A primary mechanism for block leading to reentry is nonuniform dispersion of refractoriness. A study by Geizer et al reported that a series of premature stimuli that induced large spatial dispersion of repolarization caused VF in an in vivo dog model.7 This study and others have shown that block can occur even in normal, homogeneous tissue when alternans is caused in APD. Dispersion in APD leads to the development of conduction block and to fractionation of wave fronts, which, in turn, may lead to reentrant circuits and the initiation of VT and VF (Figure 48-3). Figure 48-3 The formation of a reentrant circuit due to heterogeneity of APD Simulations have shown that the restitution curve (the relationship between APD and the diastolic interval) is predictive of the breakdown of a rotor to multiple unstable reentrant circuits. When the slope of the restitution curve (APD/diastolic interval [DI]) is greater than 1, an unstable positive-feedback loop results in increasing oscillations in APD and DI (Figure 48-4). Once the APD and the DI become so shortened that conduction is not possible, the wave front blocks and reentry may occur. A slope of less than 1 of the restitution curve leads to a negative-feedback loop, decreasing alternans, and to convergence of cycle length and DI to an equilibrium. Figure 48-4 Progression of alternans in APD and DI to conduction block and spiral wave breakdown depends on the slope of the restitution curve
Mechanisms of Ventricular Tachycardia and Fibrillation
Mechanisms of VT Onset and Maintenance
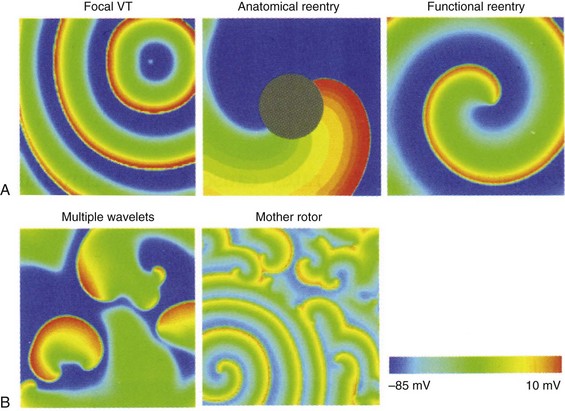
A, VT may be maintained by a focal source, around an anatomic anchor, or as a reentrant circuit around a functional anchor. B, VF may be maintained by multiple wavelet reentry or by a mother rotor (bottom left of panel) that fractionates and breaks up into multiple daughter wavelets. (Reproduced with permission from Qu Z, Weiss JN: Nonlinear dynamics of excitation and propagation in cardiac muscle. In Zipes DP, Jalife J [eds]: Cardiac Electrophysiology: From Cell to Bedside. Philadelphia, Elsevier, 2009, pp 339-348.)
Transition From VT to VF
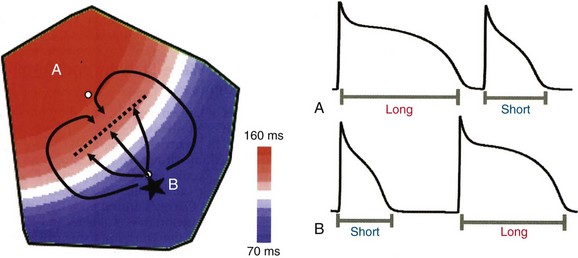
An ectopic focus at B initiates in the region of short APD and propagates outward toward the region of long APD. The ectopic beat blocks (dotted line) as it encounters refractory tissue but continues to propagate laterally such that it wraps around the area of block and reenters through the previous region of block and back into the short APD region. (Reproduced with permission from Qu Z, Weiss JN: Nonlinear dynamics of excitation and propagation in cardiac muscle. In Zipes DP, Jalife J [eds]: Cardiac Electrophysiology: From Cell to Bedside. Philadelphia, Elsevier, 2009, pp 339-348.)
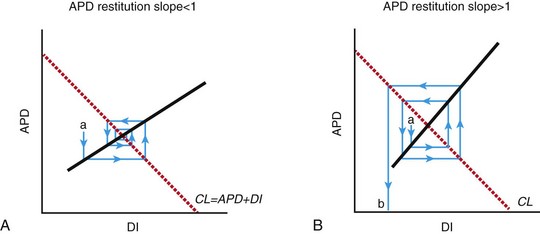
A, A spiral wave rotating at a constant cycle length (CL) undergoing a perturbation (a) that decreases DI and will tend to cause a smaller change in APD, which in turn will cause a smaller change in DI. This negative-feedback loop will eventually lead to a return to equilibrium. B, When the restitution slope >1, a perturbation in APD (a) leads to a larger disturbance in DI, which leads to a larger change in APD. This positive-feedback loop leads to increased variation in APD and DI until conduction is no longer sustained, but blocks (b). This leads to spiral wave breakup and chaotic conduction. (Reproduced with permission from Weiss JN, Garfinkel A, Karagueuzian HS, et al: Chaos and the transition to ventricular fibrillation: A new approach to antiarrhythmic drug evaluation. Circulation 99:2819-2826, 1999.)
Thoracic Key
Fastest Thoracic Insight Engine



